




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comparative study of Mo2Ga2C with superconducting MAX phase Mo2GaC: First-principles calculations
Cite this Article
Ali M A, Khatun M R, Jahan N, Hossain M M. Comparative study of Mo2Ga2C with superconducting MAX phase Mo2GaC: First-principles calculations. Chinese Physics B, 2017, 26(3): 033102 
Permissions
Comparative study of Mo2Ga2C with superconducting MAX phase Mo2GaC: First-principles calculations
† Corresponding author. E-mail:
Abstract
The structural, electronic, optical and thermodynamic properties of Mo2Ga2C are investigated using density functional theory (DFT) within the generalized gradient approximation (GGA). The optimized crystal structure is obtained and the lattice parameters are compared with available experimental data. The electronic density of states (DOS) is calculated and analyzed. The metallic behavior for the compound is confirmed and the value of DOS at Fermi level is 4.2 states per unit cell per eV. Technologically important optical parameters (e.g., dielectric function, refractive index, absorption coefficient, photo conductivity, reflectivity, and loss function) are calculated for the first time. The study of dielectric constant (ɛ1) indicates the Drude-like behavior. The absorption and conductivity spectra suggest that the compound is metallic. The reflectance spectrum shows that this compound has the potential to be used as a solar reflector. The thermodynamic properties such as the temperature and pressure dependent bulk modulus, Debye temperature, specific heats, and thermal expansion coefficient of Mo2Ga2C MAX phase are derived from the quasi-harmonic Debye model with phononic effect also for the first time. Analysis of Tc expression using available parameter values (DOS, Debye temperature, atomic mass, etc.) suggests that the compound is less likely to be superconductor.
Keyword:first-principles calculations;density of states (DOS);optical properties;thermodynamic properties
1. Introduction
Recently, the scientific community has paid significant attention to an unusual class of layered ternary carbide and nitride, the so-called MAX phases, because of their outstanding combination of properties, some of which are like ceramics and the others metallic.[1,2] To be specific, the properties with which a metal is applicable on an industrial scale are the machinability, damage tolarance, thermal and electrical conductivity, which are possessed by these materials. They also possess the properties of ceramics such as high elastic stiffness, refractory nature, and resistant to high-temperature oxidation.[3] The outstanding combination of these properties makes them attractive for potential applications in diverse fields from defense materials to electronic devices such as in defense, aerospace, automobiles, medical application, nuclear reactors and portable electronic devices where they are already used. The charismatic uniqueness of the MAX phases is motivating numerous research studies to expect that they can open the way to practical commercial applications for these materials in the future. So far, more than 70 different MAX phases have been experimentally synthesized[4] and also a good number of MAX phases have been theoretically predicted. The research in searching the new MAX phases is growing fast in order to discover more new MAX phase compounds due to their properties mentioned above. The M2AX (211) phases including solid solutions with 

Studies of Mo2Ga2C phase have been reported in the literature. The structural and compositional analysis has been addressed by Lai et al.[14] Elastic and electronic properties have been investigated by Hadi.[15] Another plausible metastable structure with close-packed Ga layers is predicted from density functional calculations by Wang et al.[16] However, though structural, elastic and electronic properties were studied, the thermodynamic and optical properties were not taken into account. Moreover, due to the similarity of structure and electronic bonding to those of superconducting Mo2GaC, Mo2Ga2C might also be a superconductor. The possibility of this property is not taken into consideration in the previous studies.
The thermodynamic properties are very important in solid state science and considered as the basis for industrial application of solids because material behavior can be obtained from thermodynamic properties under high temperatures and high pressure. Moreover, the optical properties provide the information about the electronic response of the materials which are related to the electronic properties of solids.[17] Therefore, an investigation of these properties is significantly necessary for fundamental physics and potential applications.
In this work, we aim to provide some additional information to the existing data on the physical properties of Mo2Ga2C phase by using the first-principles method, and we especially focus on the possible occurrences of superconductivity, thermodynamics, and optical properties.
2. Computational methods
The calculations were carried out by using the Cambridge Serial Total Energy Package (CASTEP) code[18] based on the density-functional theory.[19] The generalized gradient approximation (GGA) of Perdew–Burke–Ernzerhof (PBE) scheme[20] is used as the exchange and correlation function. The electrostatic interaction between valence electron and ionic core is represented by the ultrasoft pseudopotentials, and the cutoff energy for the plane wave expansion is 550 eV. A 
3. Results and discussion
3.1. Structural properties
Like all the other MAX phases, the new compound Mo2Ga2C crystallizes in the hexagonal system with space group 
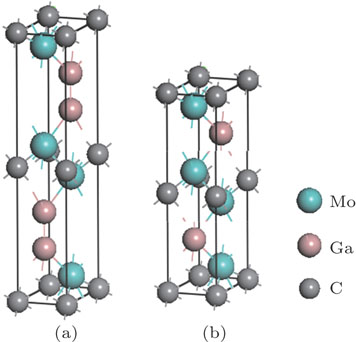
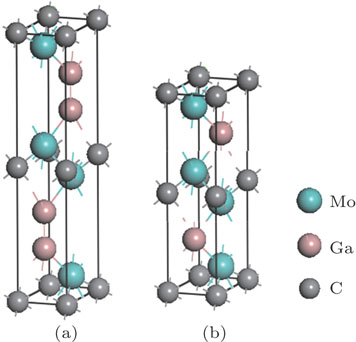 | Fig. 1. (color online) Unit cell structures of (a) Mo2Ga2C and (b) Mo2GaC. |
| Tab1e 1.
Lattice constants and atomic fractional coordinates of Mo2Ga2C and Mo2GaC. . |
3.2. Electron DOS: possibility of superconductivity
Figure 





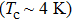
 | Fig. 2. (color online) Total and partial density of states (DOS) of (a) Mo2Ga2C and (b) Mo2GaC. |
3.3. Optical properties
Optical properties are used to describe the behaviors of materials subjected to electromagnetic radiation. In order to describe the response of Mo2Ga2C to electromagnetic radiation we calculate some important optical constants for the first time. The methods by which the optical constants are calculated can be found elsewhere.[23,25]
The optical constants of Mo2Ga2C for (100) polarization direction are shown in Fig.
The imaginary part, ε2(ω) of the dielectric function ε(ω), dominates the electronic properties of crystalline material, which depicts the probability of photon absorption. The peaks of ε2(ω) are associated with electron excitation. There is only one prominent peak around 2.0 eV (Fig.
 | Fig. 3. (a) Real part and (b) imaginary part of dielectric constant, ε1 and ε2, respectively; (c) refractive index n, (d) absorption coefficients α, (e) conductivity σ of Mo2Ga2C. |
Figure
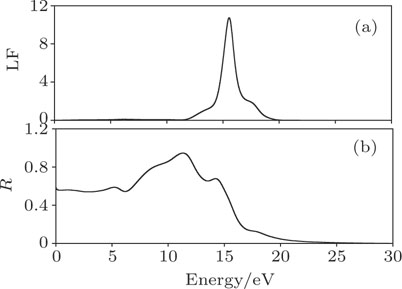
The loss function L(ω), defined as the energy loss of an electron with high velocity passing through the material as shown in Fig. 
 | Fig. 4. (a) Loss function and (b) reflectivity of Mo2Ga2C. |
The reflectivity curve is also shown in Fig.
3.4. Thermodynamic properties
The study of thermodynamic properties of materials permits a more in-depth understanding of the specific behavior of material under high temperature and pressure. The thermodynamic properties of Mo2Ga2C have been investigated by using quasi-harmonic Debye approximation.[28,29] The data calculated by using this method are in good agreement with experimental data proved by several authors.[30,31] The temperature- (0–1000 K) and pressure- (0–50 GPa) dependent polycrystalline aggregate properties including bulk modulus, Debye temperature, specific heats, and thermal expansion coefficients of Mo2Ga2C are calculated for the first time. The volume and total energy of Mo2Ga2C, calculated by the methodology described in Section
The bulk modulus, B at 0 GPa of Mo2Ga2C as a function of temperature is shown in Fig.
 | Fig. 5. Temperature-dependent (a) bulk modulus, B and (b) Debye temperature, ΘD of Mo2Ga2C. The insets show pressure dependence. |
Figure 
 | Fig. 6. Temperature dependence of lattice specific heat at constant volume (a) and specific heat at constant pressure (b) of Mo2Ga2C. |
The lattice heat capacity of a substance is a measure of how well the substance stores heat. The temperature dependence of CV is governed by the details of vibrations of the atoms and could be determined from experiments. It is worthwhile to outline that the Debye model correctly predicts the low-temperature dependence of the heat capacity at constant volume, which is proportional to T3.[33] It also recovers the Dulong–Petit law at high temperature.[34] The heat capacities at constant-volume (CV) and constant-pressure (CP) of Mo2Ga2C each as a function of temperature are displayed in Figs. 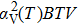

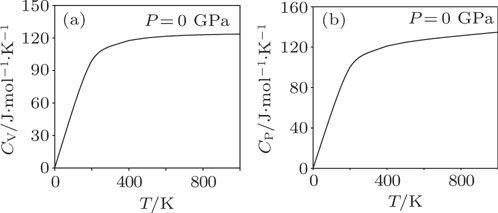
The volume thermal expansion coefficient, αV, as a function of temperature and pressure is shown in Fig. 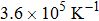


 | Fig. 7. Plot of volume thermal expansion coefficient versus temperature of Mo2Ga2C. The inset shows the pressure dependence. |
4. Conclusions
The optical and thermodynamic properties of Mo2Ga2C and the prediction of the occurrence of superconductivity are investigated for the first time by the first-principles method. The thermodynamic properties are derived from the quasi-harmonic Debye model with phononic effect. The energy bands around the Fermi level are mainly from Mo 4d states, suggesting that the Mo 4d states dominate the conductivity. The analysis of the electronic band structure indicates the metallic behavior of the compound, which is also confirmed by the studies of absorption and conductivity spectra. All optical functions are calculated in the polarization direction (100) and analyzed in detail. The results are in good agreement with other reported results of Mo2Ga2C. The results are also compared with those of the Mo2GaC, which are available. Based on our present study, it is worthwhile to say that the Mo2Ga2C material could be used as a technologically important material.
Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] | |
| [32] | |
| [33] | |
| [34] |