



{kind=link}
{kind=link}
{kind=link}
{kind=link}
First-principle investigation on perovskite La1−x EuxGaO3
[Gu Yanni1, 2, Xu Sheng1, 2, Wu Xiaoshan1, †,  ]
]
]
|
|


† Corresponding author. E-mail:
Project supported by the National Natural Science Foundation of China (Grant Nos. U1332205, 11274153, 10974081, and 10979017) and the Postdoctoral Fund of Jiangsu Province, China (Grant No. 1301019B).
The pseudopotential method has been used to investigate the structural, electronic and magnetic properties of La1−xEuxGaO3 (x = 0, 0.25, 0.5, 0.75, and 1) within the scheme of generalized gradient approximation. The spin-polarized calculations demonstrate that the ground state is an antiferromagnetic insulator for x ≤ 0.5, while it is ferromagnetic half-metal at x > 0.5. The substitutions of magnetic Eu ions for non-magnetic La ions produce and strength spin polarization, which forcefully urges the system from the insulator to the half metal. Meanwhile, Eu doping strengthens a stoner mechanism for ferromagnetism of La1−xEuxGaO3 (x = 0.75 and 1), which may lead to a rapid increasing in the total magnetic moment and therefore, antiferromagnetic–ferromagnetic transition happens.
Many researchers persistently focus substantial concern on orthogallates due to their particular physical properties and promising applications in devices. LaGaO3 is one of these compositions. It is the most suitable compound for interconnecting material in solid oxide fuel cells (SOFC).[1–5] Aside from possible applications for electrolyte of solid fuel, LaGaO3 has many other potential applications, such as luminescence and thermal potential.[6–10]
At room temperature, LaGaO3 crystal has an ABO3 perovskite structure with GdFeO3-type distortion and falls into the orthorhombical space group Pbnm.[11] GaO6 octahedra are not aligned along the unit-cell axes. Lanthanide ions doping on A-sites have significant effects on the structural and physical properties of LaGaO3. Liu et al.[6–9] reported Ln3+-doped (Ln = Eu, Dy, Tm, Tb, Sm, and Tb) LaGaO3 nanocrystalline phosphors, investigating the crystallization process, photoluminescence, and cathodoluminescence properties. Parveen et al. have studied the thermal properties of La1−xLnxGaO3 (Ln = La, Ce, Nd, Pr).[10] Sood et al. studied structural and electronic behavior of Ba-doped LaGaO3.[12]
Most of these researches are focused experimentally on the photoluminescence, cathodoluminescence and thermal properties, etc., while there is almost no theoretical work on electronic structure and magnetic property for Ln3+-doped LaGaO3 (Ln = rare earth elements) although there are some first-principle researches about effects of doping on physical properties of other systems within density-functional theory.[13–17] Theory research has important instruction significance for experimental work.
Here, the investigation of structural, electronic, and magnetic properties for La1−xEuxGaO3 (0 ≤ x ≤ 1) are carried out using the generalized gradient approximation (GGA). The unit cell stability, the electronic structure, and the magnetic structure are determined. The further experiments are needed to confirm the present transition of La1−xEuxGaO3 from antiferromagnetic (AFM) insulator to ferromagnetic (FM) half metal.
We performed the first-principle calculations of La1−xEuxGaO3 (x = 0, 0.25, 0.5, 0.75, and 1) using the GGA method and the projector-augmented wave (PAW)[18] potentials, which are implemented in the Vienna ab initio Simulation Package (VASP)[19,20] program. In order to study the structural, electronic and magnetic properties of La1−xEuxGaO3, a supercell has been constructed consisting of 40 atoms with size 
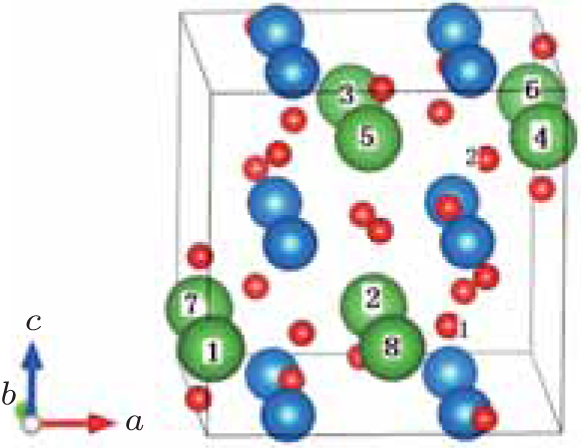 | Fig. 1. A 40-atom supercell of perovskite structure LaGaO3. The green, blue, and red spheres represent La, Ga, and O atoms, respectively. Eight different sites of La atoms and two O atoms are marked with arabic numerals. |
The calculated structural parameters for different compositions are given in Table
| Table 1. Calculated structural parameters and total magnetic moments for La1−xEuxGaO3 (x = 0, 0.25, 0.5, 0.75, 1). . |
The cell volume, bond distances d(Eu1−O1) and d(La5−O2) decrease linearly with the increasing Eu composition x, as shown in Fig.
 | Fig. 2. The composition x dependence of calculated unit cell volume V in La1−xEuxGaO3. |
The GGA calculation results show that the electronic ground states of this system are insulators when x ≤ 0.5, and are half-metals for x > 0.5. The calculated total density of states (TDOS) of La1−xEuxGaO3 and partial Eu, Ga, O, and La atomic projected DOS (PDOS) for different contents are shown in Fig.
 | Fig. 3. The total DOS and projected DOS of La1−xEuxGaO3 (x = 0, 0.25, 0.5, 0.75, 1). Fermi levels are black vertical lines in the figure. |
 | Fig. 4. Band decomposed charge density of (a) value band at x = 0, (b) conduction band at x = 0, (c) value band at x = 0.25, (d) conduction band at x = 0.25, (e) value band at x = 0.5, (f) conduction band at x = 0.5, (g) bands near Fermi level at x = 0.75, and (h) bands near Fermi level at x = 1. We show the bands near Fermi level at x = 0.75 and x = 1 because these materials are half-metal according to the present results. |
LaGaO3 is known as an insulator, which corresponds with our DOS calculation (see Fig.
For x = 0.25 in La1−xEuxGaO3 (see Fig.
For x = 0.5 (see Fig.
| Table 2. The atoms in the parentheses show the sites where La atoms were replaced. The third and last columns show the magnetic moment at various inequivalent sites. . |
With further increasing of Eu concentration, the DOS of the compounds change inevitably. For x = 0.75 (see Fig.
Finally, our calculation results demonstrate that EuGaO3, i.e., the compound x = 1 (see Fig.
In summary, DOS and PDOS of LaGaO3 are symmetric between spin-up and spin-down bands as shown in Fig.
The total magnetic moments and atom moment for La1−xEuxGaO3 are listed in Table
The magnetic structure of La0.5Eu0.5GaO3 is also an AFM state where each moment for Eu ions is aligned antiparallel with the nearest-neighbor one. For La0.25Eu0.75GaO3 and EuGaO3, the FM configuration has the lowest energy and is assumed to be the ground state. The FM state of EuGaO3 is 0.26 eV lower than the G-AFM state. We tested three types of AFM structures of EuGaO3 and found that the G-type AFM is 0.12 eV and 0.30 eV lower than the A-type and C-type AFM, separately.
In the La-rich regime, La1−xEuxGaO3 (0 < x ≤ 0.5) is AFM so that the total magnetic moments (μT) equal to 0. As the Eu content keeps increasing, we find a rapid increasing in μT. La0.25Eu0.75GaO3 and EuGaO3 have an FM structure. As localized Eu 4f electrons increase, the hybridization of Eu (4f)–O (2p) is strengthened, which strengthens the exchange splitting, and increases the spin magnetization of the system. The increase in the exchange splitting shows Eu doping strengthens the effect of the Stoner mechanism for ferromagnetism in La1−xEuxGaO3 (x > 0.5), which may lead to a rapid increasing in μT. So La1−xEuxGaO3 (x > 0) compounds change from an AFM state to an FM state with x increasing.
In short, we studied the electronic structure and magnetic properties of the La1−xEuxGaO3 with x of 0, 0.25, 0.5, 0.75, and 1 within the GGA scheme. The crystal structures of the compounds have perovskite structures. The volumes over the range of compounds (0 ≤ x ≤ 1) decrease with increasing x. The calculations results demonstrate that La1−xEuxGaO3 is an insulator ground state for the x = 0, 0.25 and 0.5 compounds and a half-metallic ground state for the x > 0.5 regimes. The magnetic structure for 0 < x ≤ 0.5 is AFM. The magnetic structure for x > 0.5 is found to be ferromagnetic. The substitution of non-magnetic La ions by magnetic Eu ions drives the system from AFM insulator to the FM half metal.
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 | |
| 22 | |
| 23 | |
| 24 | |
| 25 | |
| 26 | |
| 27 | |
| 28 | |
| 29 |