{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Thermophysical properties of iridium at finite temperature
[Kumar Priyank1, †,  , Bhatt N K2, Vyas P R3, Gohel V B3]
, Bhatt N K2, Vyas P R3, Gohel V B3]
, Bhatt N K2, Vyas P R3, Gohel V B3]
|
|


† Corresponding author. E-mail:
Project supported by the Department of Science and Technology–Fund for Improvement of Science and Technology Infrastructure Project (DST-FIST) (Level 1) of Department of Sciences and Technology (DST), New Delhi (Grant No. SR/FST/PST-001/2006).
The bulk properties of materials in an extreme environment such as high temperature and high pressure can be understood by studying anharmonic effects due to the vibration of lattice ions and thermally excited electrons. In this spirit, in the present paper, anharmonic effects are studied by using the recently proposed mean-field potential (MFP) approach and Mermin functional which arise due to the vibration of lattice ions and thermally excited electrons, respectively. The MFP experienced by a wanderer atom in the presence of surrounding atoms is constructed in terms of cold energy using the local form of the pseudopotential. We have calculated the temperature variation of several thermophysical properties in an extreme environment up to melting temperature. The results of our calculations are in excellent agreement with the experimental findings as well as the theoretical results obtained by using first principle methods. We conclude that presently used conjunction scheme (MFP+pseudopotential) is simple computationally, transparent physically, and accurate in the sense that the results generated are comparable and sometimes better than the results obtained by first principle methods. Local pseudopotential used is transferable to extreme environment without adjusting its parameters.
Iridium (Ir) is a 5d transition metal of platinum group with a high density of d-states at the Fermi level which is the main hindrance in the theoretical investigation of its lattice properties. It is the second densest metal after osmium and is found in face centred cubic (fcc) structures up to melting temperature. It is different from other fcc transition metals due to some of its unusual physical properties, such as high melting temperature, high electrical conductivity, large shear and bulk moduli and negative Cauchy pressure. It is very hard, brittle and corrosion resistant and its phonon spectra show Kohn anomalies.[1] Its thermal stability at high temperature and the large shear modulus make it a superior metal for ultra high temperature applications. Physical properties of Ir at high temperature and high pressure are not yet completely known. To understand the behavior of these materials in an extreme environment (high temperature and high pressure), one requires knowledge of thermophysical properties. Such study provides important information about the new materials which are needed in modern technological applications. At high temperature, thermal vibrations (anharmonic effects (AE)) play an important role in the determination of thermophysical properties of materials. At this temperature (near melting temperature), the contribution due to vacancies and thermally excited lattice defects to the physical properties is noticeable. Hence, it is difficult to know the magnitude of contribution due to AE from the above mentioned contributions, which leads to large uncertainties in the study of physical properties of materials at such high temperatures experimentally.[2] First principle calculations are highly reliable and they are a better supplement of experimental methods for the study of thermophysical properties of materials in an extreme environment, but the study of phonon contribution to thermodynamic properties of materials is beyond the capability of ab-initio approach because it is too complex mathematically and computationally lengthy which is implemented by using certain well defined approximations. In comparison with first principle methods, the other methods based on theoretical modeling are computationally simple and physically transparent and they can explain a wide range of physical properties of different groups of materials not only qualitatively but also quantitatively.
Very recently, many attempts have successfully been made to theoretically investigate the static, lattice dynamical and thermodynamic properties of Ir using first principle methods[3,4] and other methods[5,6] including pseudopotential.[1,2,7–9] All these studies were carried out to theoretically understand the behavior of microscopic properties of materials. However, limited attempts have been made to understand the behavior of materials at extreme condition from macroscopic properties.[10,11] This fact encouraged us to study the thermophysical properties of Ir at high temperatures.
Recently, Wang and his co-workers[12–16] proposed a mean-field potential (MFP) approach to account for the vibrational contribution of the lattice ions to the total free energy, which means that the field potential is defined as the potential experienced by a wanderer atom in presence of surrounding atoms which is computed in terms of cold energy. These authors have accounted for the contribution due to thermally excited electrons by computing energy density of states.
In this paper, by using a simple scheme proposed by Bhatt and co-workers,[17–22] we have studied vibrational contribution to the free energy using mean-field potential approach in conjunction with Krasko–Gurskii (KG)[23] pseudopotential. The contribution to the free energy due to thermally excited electrons has been accounted for by using Mermin functional.[24] We have studied volume variation of cold energy (Ec) as well as Helmholtz free energy (Fion(Ω, T)) at different temperatures along with the equation of state (EOS) at 300 K and shock Hugoniot. Thermal expansion (βP) isothermal and adiabatic bulk moduli (BT and BS), specific heats at constant pressure (CP) and constant volume (CV) and enthalpy (EH) have also been studied in this paper.
The rest of this paper is arranged as follows. In Section 2, we describe the mathematical methods to compute physical properties. In Section 3, our computed results are compared with the available experimental findings and theoretical results. Section 4 is concluding remarks.
The total Helmholtz free energy per atom is given by[12]
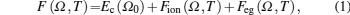
The unscreened KG pseudopotential[23] has the following form in q space:
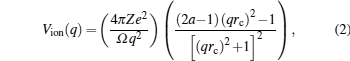
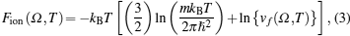
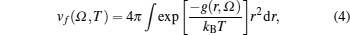
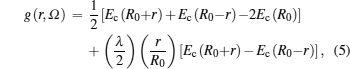
The free energy due to thermal excitation of a free electron gas (Feg), can be written using Mermin functional[24] as
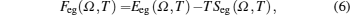
The first step of our calculation is to determine the pseudopotential parameters (a and rc) appearing in Eq. (
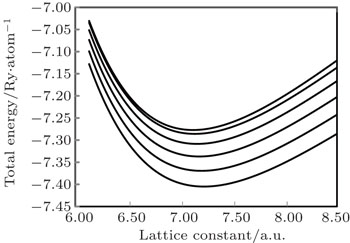
In the present study, the experimental binding energy has been calculated as the sum of experimental values of cohesive energy and ionization energies due to valence electrons,[29,30] which were taken from Refs. [31] and [32], respectively. Our calculated binding energy is in excellent agreement with the experimental result and the calculated lattice constant has 1.77% variation with the experimental result reported in Ref. [3]. The excellent agreement of the computed binding energy with the experimental result suggests that such energy, known as intermolecular energy which is responsible for holding the individual components of metal together, is properly accounted for. Park et al.[3] have used van der Waals (vdW) interaction with standard density functional theory (DFT). In their computation, they used five van der Waals density functional (vdW-DF) and five dispersion corrected (DFT-D) vdW functional along with the standard DFT functional of local density approximation (LDA) and generalized gradient approximation (GGA). In the present study, the lattice constant at 0 K has been computed from the minima of static-energy versus the volume curve which is shown in Fig.
| Table 1. The comparison of computed lattice constant (R0 in a.u.), binding energy (−E in Ry/atom), and static bulk modulus (B0 in GPa) with experimental and other theoretical results. . |
The Helmholtz free energy as a function of temperature is computed by using Eq. (
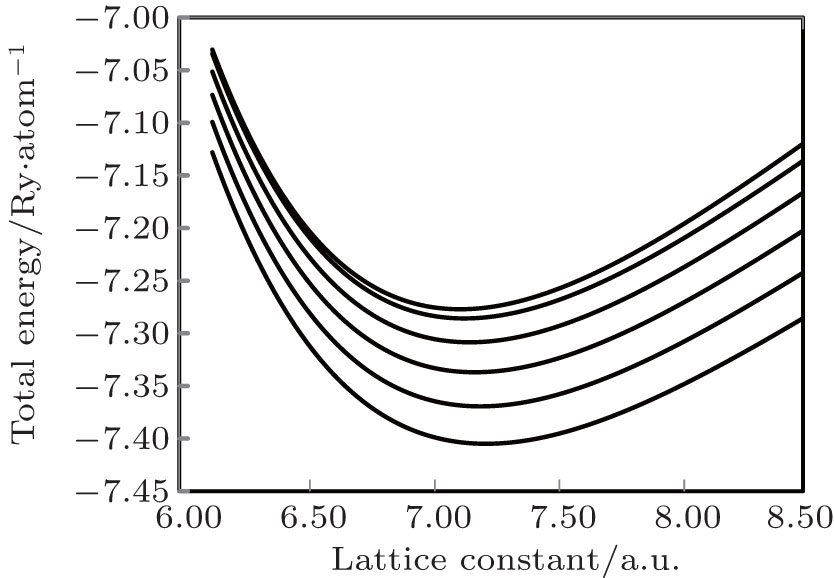 | Fig. 1. The volume variations of cold energy and Helmholtz free energy. From top to bottom graphs, the curves correspond to temperatures: T = 0 K, 500 K, 1000 K, 1500 K, 2000 K, and 2500 K, respectively. |
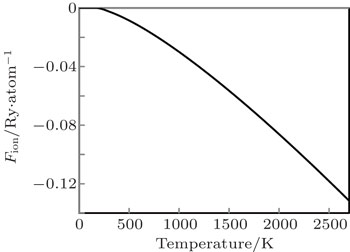
The variation of free energy due to lattice ion (Fion) against temperature is shown in Fig.
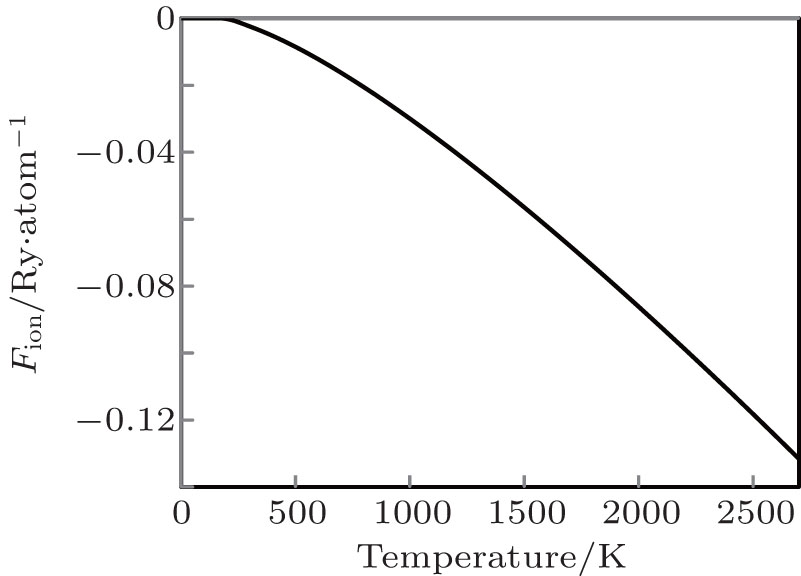 | Fig. 2. The variation of free energy due to lattice ion (Fion) against temperature. |
The behavior of the expansion of material with temperature can be understood by studying coefficient of volume-thermal expansion (βP) which is defined as[12]
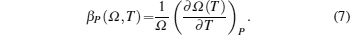
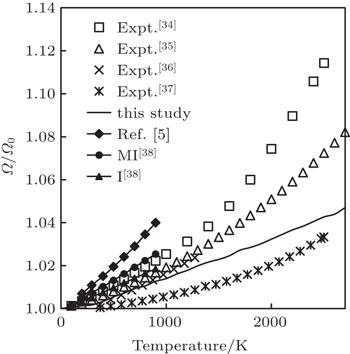
During the literature survey, we observed that different authors have reported the values of linear, volume and relative volume thermal expansions (Ω/Ω0, where Ω and Ω0 are volumes at given temperature and 0 K, respectively). In order to compare our computed results of volume thermal expansion with the experimental and other theoretical results, we have converted the results in the form of relative volume thermal expansion. The temperature variation of relative volume thermal expansion along with the experimental findings[34–37] and other theoretical results[5,38] is shown in Fig.
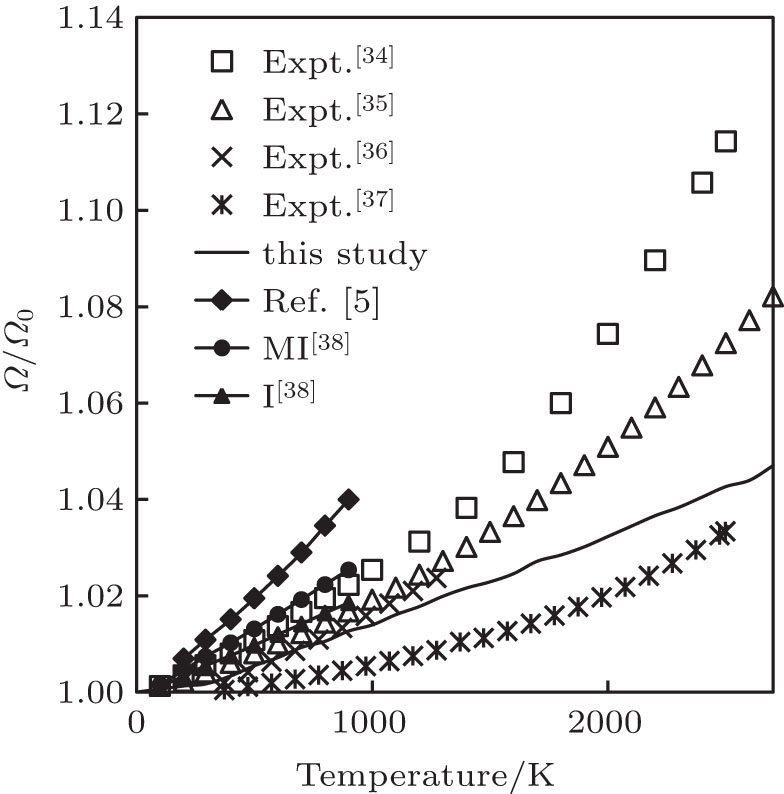 | Fig. 3. Relative volume thermal expansion as a function of temperature. Here, “I” represents pseudopotential of Ivanov, while “MI” represents the modified form of the pseudopotential of Ivanov. |
To the best of our knowledge, for the first time, in this paper, we have carried out the study of thermal expansion of Ir up to melting temperature because, at higher temperatures, the experimental results of βP change appreciably and the theoretical studies are limited up to 900 K. By conducting a detailed comparison of our results with experimental results, we find that our results are almost identical to experimental results obtained by Touloukian et al. (recommended values) up to 1500 K.[34] The maximum deviation of our computed results with the results obtained by Touloukian et al. is 6.9% at 2500 K. The results obtained by Arblaster[35] are also in good agreement with 2.78% variation at 2500 K. The results obtained by Caldwell[36] are in excellent agreement with our computed results up to 1300 K. The results obtained by Halvorson[37] are in excellent agreement with our theoretical results at low temperature and high temperature with maximum deviation of 1.05% at 1800 K. Our computed results are also better than the theoretical results obtained by Ferah et al.[5] and Katsnelson et al.[38] Ferah et al.[5] used molecular dynamics simulations by using modified Morse potential and employed embedded atom method (EAM) to study temperature variation of thermal expansion. Katsnelson et al.[38] used pseudopotential obtained by Ivanov et al. (I) and modified form of the pseudopotential of Ivanov et al. (MI) to study the temperature variation of thermal expansion. From such detailed comparisons, we find that the simple method used in this paper properly accounts for anharmonic effects.
The EOS which is a fundamental property of material plays a crucial role in understanding a wide range of physical properties of material in condensed matter physics. For modeling the Earth’s core and planetary interiors, it is one of the important parameters. EOS is very useful in geophysics, astrophysics, and material science. Static EOS at ambient temperature which is largely governed by cold energy provides a link to monitor microscopic internal structure of the material. 300 K EOS has been computed by using the following equation:
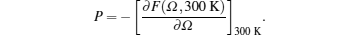
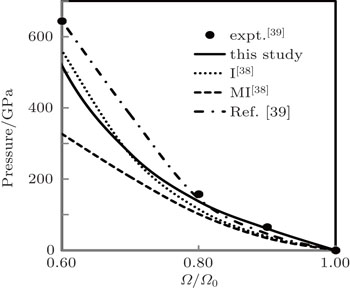
Our computed P–Ω isotherm at 300 K is compared with experimental findings[39] as well as theoretical results[38,39] in Fig.
 | Fig. 4. Our computed P–Ω isotherm at 300 K compared with experimental findings[39] as well as theoretical results[38,39] up to 40% compression. |
Our results are in excellent agreement with the results obtained by Katsnelson et al.[38] using I for all compression range and in excellent agreement for smaller compressions up to 20% and in reasonably good agreement at higher compressions using MI. From above comparison, we find that our results are better than other theoretical results,[38,39] which confirm the applicability of local form of the pseudopotential used in the present calculation which has only one adjustable parameter rc (core radius). Further, EOS is a test of approximations used in the theory for the electron–electron interaction which seems to be properly accounted for.
The internal energy or enthalpy at zero pressure is given by[12]
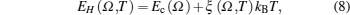
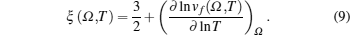
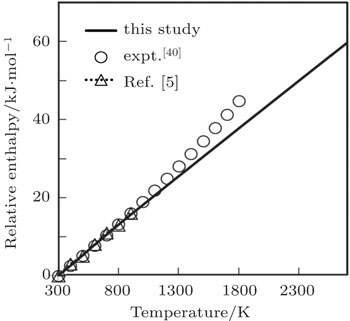
Temperature dependence of computed values of relative or scaled enthalpy (EH(T)−EH(300 K)) up to melting temperature and the experimental results up to 1800 K obtained by Robie et al.[40] and the theoretical results up to 900 K obtained by Ferah et al.[5] are compared in Fig.
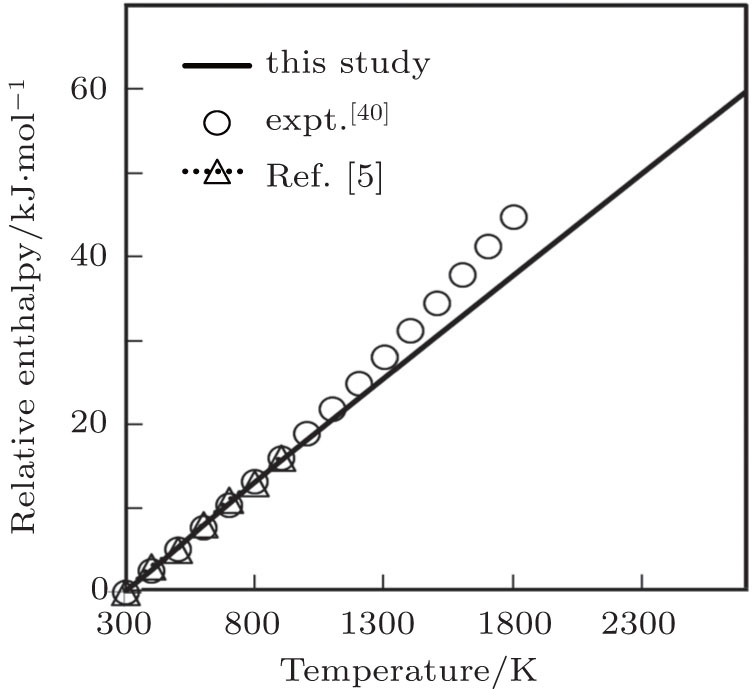 | Fig. 5. Relative enthalpy as a function of temperature. |
The isothermal (BT) and adiabatic (BS) bulk moduli are calculated using the following equations:[10]
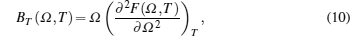
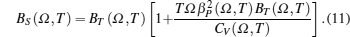
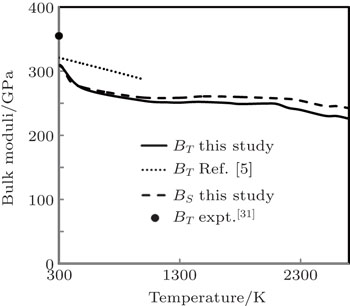
The temperature variation of our computed results of isothermal and adiabatic bulk moduli up to melting temperature and the theoretical results of BT obtained by Ferah et al.[5] (up to 1000 K) along with the experimental point of BT at 300 K[31] are displayed in Fig.
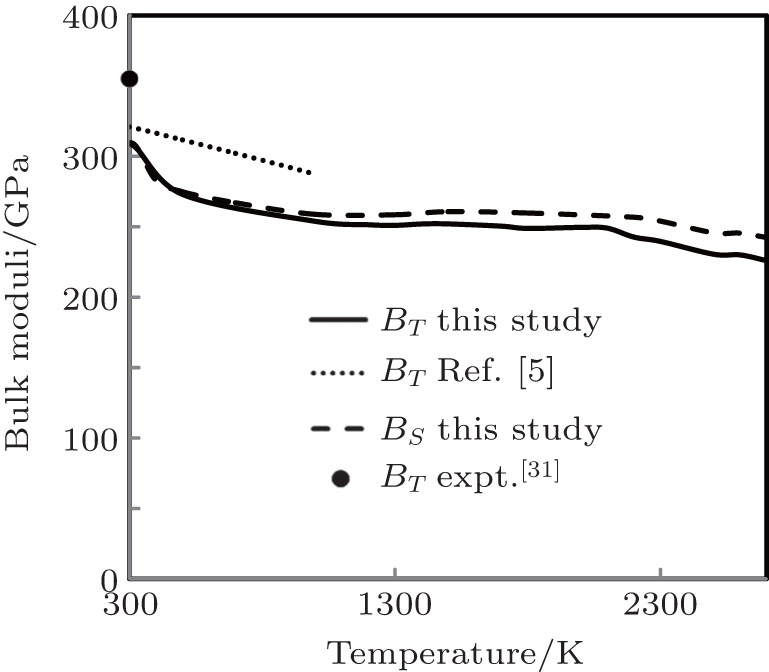 | Fig. 6. Bulk moduli as a function of temperature. |
At 300 K, the computed result of BT has 14.92% variation with the experimental value,[31] which is better than the result of BT at 0 K[3] with 30.64%variation. Such observation reveals that percentage variation of BT with the experimental result decreases with the increase in temperature. In the absence of experimental results, at higher temperature, one can say that our approach explains anharmonocity at higher temperatures in a better way. Our computed results of BT are in good agreement (having 3.71% variation at 300 K and 11.60% variation at 1000 K) with the theoretical results obtained by Ferah et al.[5] In the absence of experimental and theoretical results of BS, we could not compare it.
The total contribution of specific heat at constant volume (CV) arises due to contributions from atomic vibrations 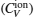
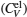
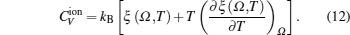
The total constant volume heat capacity (CV) and the constant pressure heat capacity (CP) are given by the following equations:[10]
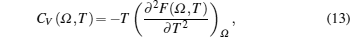
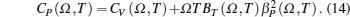
Anharmonic contribution to the heat capacity 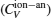
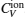
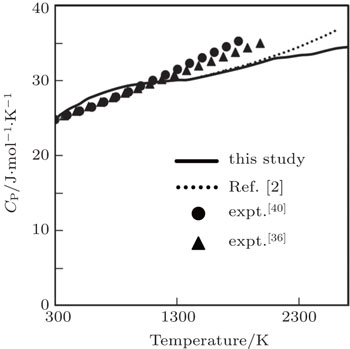
Our computed results of CP are in excellent agreement with the experimental results (up to 1800 K) obtained by Robie et al.[40] The variation increases with the increase in temperature with maximum variation of 11.60% at 1800 K. The experimental results of CP obtained by Caldwell[36] are also in excellent agreement with the present result with 7.60% variation at 2000 K. Our computed results of CP are also in excellent agreement with the theoretical results obtained by Katsnelson et al.[2] who have used pseudopotential of Animalu and Heine in the framework of second-order perturbation theory.
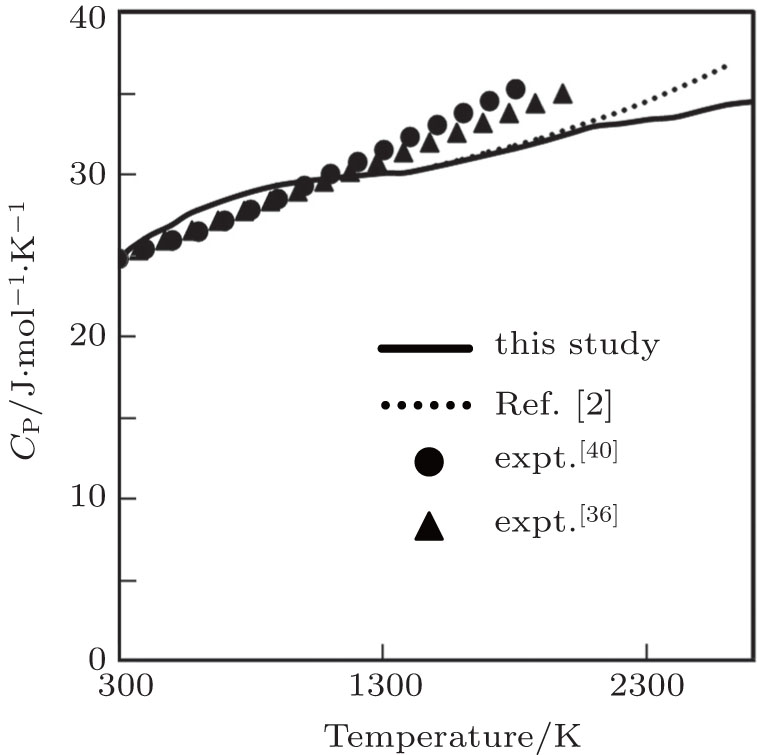 | Fig. 7. Specific heat at constant pressure as a function of temperature. |
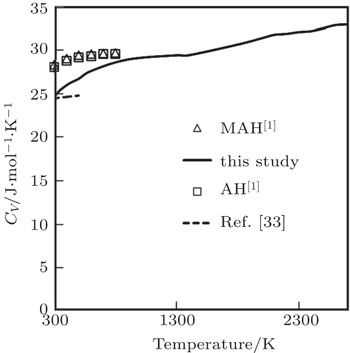
The temperature dependence of our computed results of CV along with the corresponding other theoretical results[1,33] is displayed in Fig.
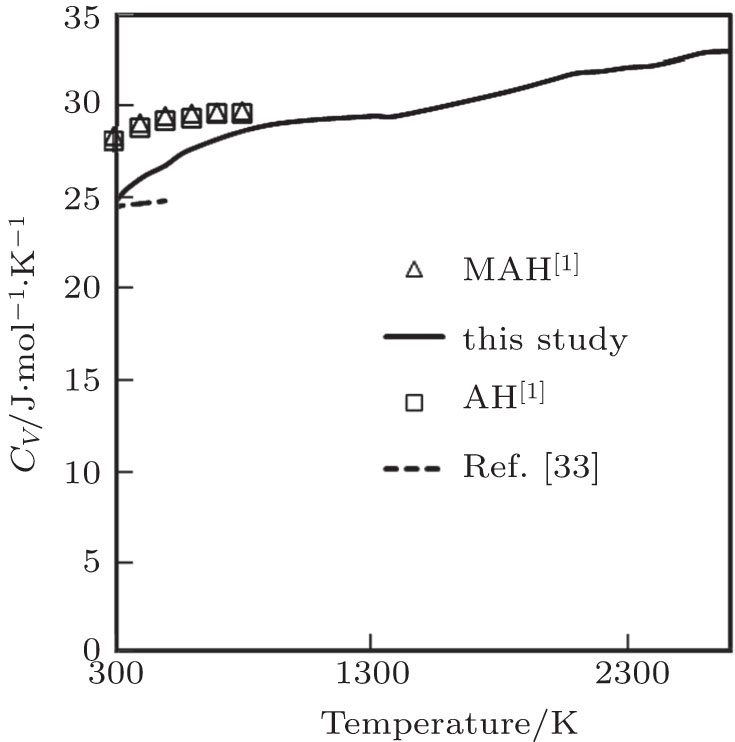 | Fig. 8. Specific heat at constant volume as a function of temperature. |
Our computed results of CV are in excellent agreement with the theoretical results obtained by Katsnelson et al.[1] and Ahmed et al.[33] Ahmed et al.[33] used a special ab-initio density functional theory along with pseudopotential plane wave (PP-PW) method, by using Vanderbilt formalism with local density approximation (LDA) for the exchange and correlation energy of electrons. Katsnelson et al.[1] studied CV by using Animalu–Heine pseudopotential (AH) and also with modification in Animalu pseudopotential (MAH). Authors[1] have used effective valency 4.5 instead of actual valency. Here, we would like to point out that during calculation, the volume variation with temperature in the calculation of CV was not considered in Ref. [1].
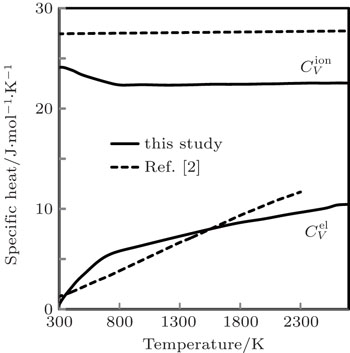
The computed results of 
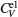
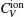
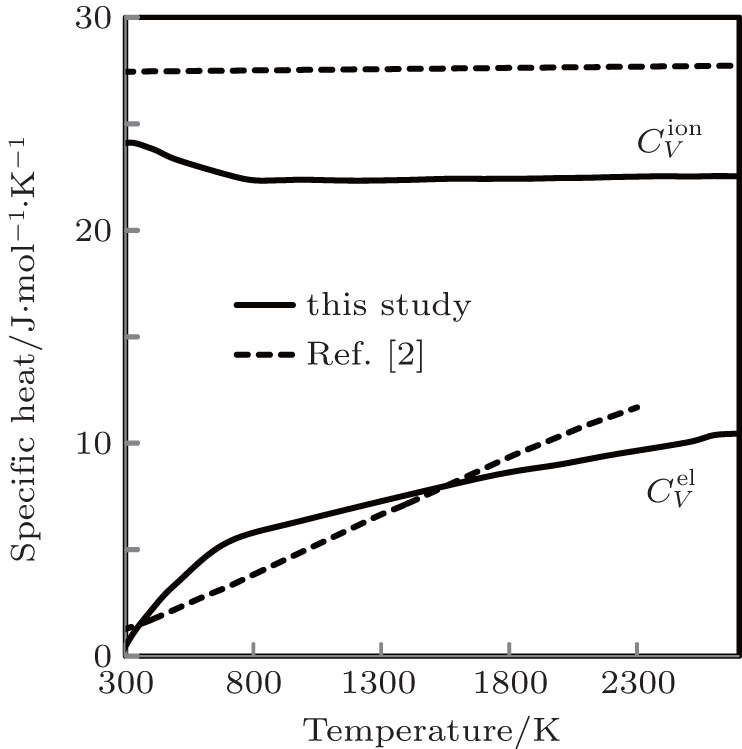 | Fig. 9. Specific heats due to lattice ions and thermally excited electrons as a function of temperature. |
In the shock-wave experiment, a supersonic wave is passed through the material which produces simultaneous high temperature and high pressure. The dynamic EOS or shock-Hugoniot or simply Hugoniot is defined as the locus of all possible states that can be reached by using a single shock from a given initial state, usually defined by thermodynamic variables like pressure, volume and internal energy or enthalpy. The Rankin–Hugoniot equation is given by
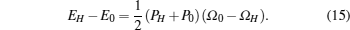
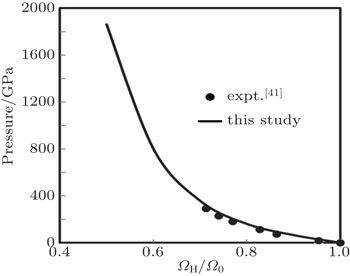
Here, PH, ΩH, and EH are pressure, volume, and energy of the compressed state while P0, Ω0 and E0 are corresponding quantity under ambient condition. Equation (
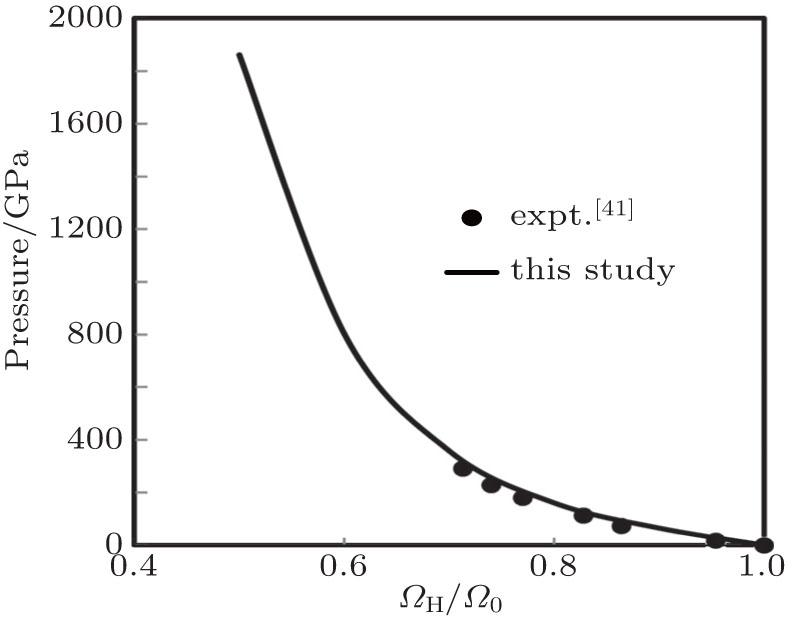 | Fig. 10. Results of Hugoniot pressure as a function of fraction of volume. |
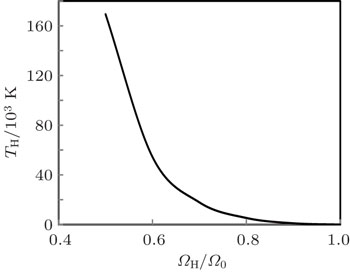
Volume variation of temperature along Hugoniot (TH) is shown in Fig.
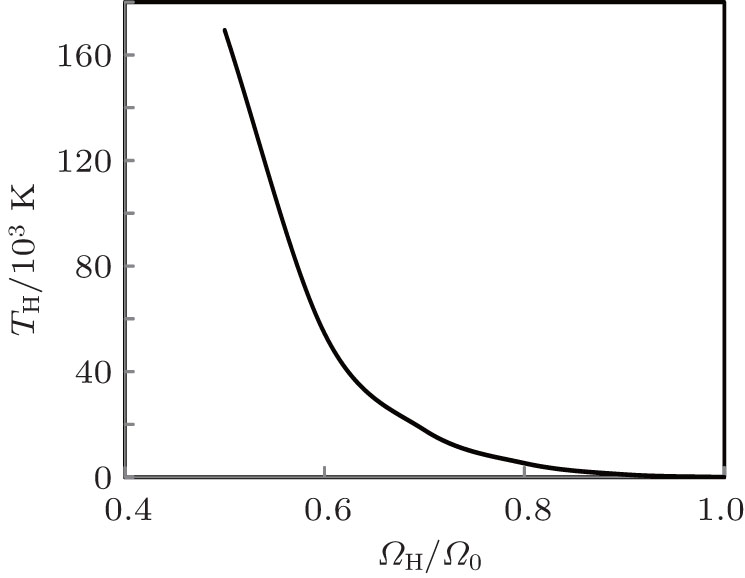 | Fig. 11. Calculated Hugoniot temperature as a function of the reduced volume. |
We have carried out a theoretical study of the thermophysical properties of Ir using a simple local form of the pseudopotential in conjunction with MFP approach at high temperature (up to melting temperature) and high pressure. The computed results of thermophysical properties at different temperatures are in excellent agreement with the experimental findings as well as the results obtained by using first principle methods. Such success reveals that the anharmonic effect which plays a vital role in the determination of thermophysical properties of materials at higher temperatures is included properly in the present conjunction scheme. One of the most important advantages of the MFP approach used in the study is that it accurately bypasses the lengthy and intricate calculations required for the calculation of Fion using the phonon density of states. In such an approach, for the theoretical calculation of the lattice constant as a function of temperature, one requires volume variation of phonon density of states at each temperature. Our results also confirm that the accurate description of the thermophysical properties at higher temperature can be done by including the effect of electron–electron interaction which can be explained by using Mermin functional instead of calculating energy density of states.[12] In our opinion, the pseudopotential approach is computationally simple and physically transparent, at the same time; it generates accurate results in an extreme environment which are comparable with first principle methods. The local form of pseudopotential (KG) with only one adjustable parameter rc (determined from zero pressure condition at 0 K) is found to be transferable without changing its value at extreme environment during calculation. Further, the present calculation is free from any kind of adjustment of valency and does not require a Born–Mayer-type potential to account core–core repulsion.[1,2,9] Moreover, as used by many researchers,[1] in the present scheme, we do not require an extra analytical form of the potential in q space which is added to pseudopotential for the better description of many thermodynamic properties. In that sense, s–p–d hybridization is accounted for properly in the pseudopotential itself.
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 | |
| 22 | |
| 23 | |
| 24 | |
| 25 | |
| 26 | |
| 27 | |
| 28 | |
| 29 | |
| 30 | |
| 31 | |
| 32 | |
| 33 | |
| 34 | |
| 35 | |
| 36 | |
| 37 | |
| 38 | |
| 39 | |
| 40 | |
| 41 |