Wang Jun-Fei, Fu Xiao-Nan, Zhang Xiao-Dong, Wang Jun-Tao, Li Xiao-Dong, Jiang Zhen-Yi. Structural, elastic, electronic, and thermodynamic properties of MgAgSb investigated by density functional theory. Chinese Physics B, 2016, 25(8): 086302
Permissions
Structural, elastic, electronic, and thermodynamic properties of MgAgSb investigated by density functional theory
Wang Jun-Fei1, Fu Xiao-Nan1, Zhang Xiao-Dong1, 2, †, , Wang Jun-Tao1, Li Xiao-Dong1, Jiang Zhen-Yi2
College of Science, Henan University of Technology, Zhengzhou 450001, China
Institute of Modern Physics, Northwest University, Xi’an 710069, China
Project supported by the National Natural Science Foundation of China (Grant No. 11504088), the Fund from Henan University of Technology, China (Grant Nos. 2014YWQN08 and 2013JCYJ12), the Natural Science Fund from the Henan Provincial Education Department, China (Grant No. 16A140027), the Natural Science Foundation of Shaanxi Province of China (Grant Nos. 2013JQ1018 and 15JK1759), and the Science Foundation of Northwest University of China (Grant No. 14NW23).
Abstract
Abstract
The structural, elastic, electronic, and thermodynamic properties of thermoelectric material MgAgSb in γ,β,α phases are studied with first-principles calculations based on density functional theory. The optimized lattice constants accord well with the experimental data. According to the calculated total energy of the three phases, the phase transition order is determined from α to γ phase with cooling, which is in agreement with the experimental result. The physical properties such as elastic constants, bulk modulus, shear modulus, Young’s modulus, Poisson’s ratio, and anisotropy factor are also discussed and analyzed, which indicates that the three structures are mechanically stable and each has a ductile feature. The Debye temperature is deduced from the elastic properties. The total density of states (TDOS) and partial density of states (PDOS) of the three phases are investigated. The TDOS results show that the γ phase is most stable with a pseudogap near the Fermi level, and the PDOS analysis indicates that the conduction band of the three phases is composed mostly of Mg-3s, Ag-4d, and Sb-5p. In addition, the changes of the free energy, entropy, specific heat, thermal expansion of γ-MgAgSb with temperature are obtained successfully. The obtained results above are important parameters for further experimental and theoretical tuning of doped MgAgSb as a thermoelectric material at high temperature.
The half-Heulser alloys have attracted a lot of attention because of their abundant magnetic,[1,2] thermoelectric properties,[3–6] and behaviors as tunable multifunctional topological insulators.[7] The thermoelectric properties of half-Heusler compounds can be modulated by substituting the three atomic positions. MgAgSb with 18 valence electrons has been found to have a half-Heusler structure. Moreover, the figure of merit reaches a maximum value of 0.56 at temperatures around 150 °C to 170 °C.[8] Ying et al.[9] demonstrated that α-MgAgSb is a promising candidate for power generation (300 K–550 K). Therefore, it is expected to be used as a thermoelectric material in a high temperature environment. Recently, the p-type MgAgSb-based material which shows great potential due to its high figure of merit (≈ 1.4 at 475 K) has been reported.[10] The p-type MgAgSb-based compound has been shown to have a high thermoelectric conversion efficiency of 8.5% between 20 °C and 245 °C.[11] Other strategies have been reported to improve the thermoelectric performances of MgAgSb-based materials for low-temperature power generation.[12] The crystal structure and some physical properties of MgAgSb were studied by Kirkham et al.,[8] Frost and Raynor.[13] For example, the MgAgSb exhibits three different structures in the temperature range of 30 °C–420 °C.[8]
Elastic properties determine the material brittleness and elastic response to external strain, which is very common in material processing. The knowledge of elastic properties not only strengthens the fundamental understanding of the material performance under the strain, but also can relate the electronic structures in different strained conditions. The sound velocity and Debye temperature are not only the important factors describing the fundamental physics properties of materials, but also can be used to estimate the thermal properties of different phases by using the Debye model.
For MgAgSb, despite its potential application as a thermoelectric material, many physical properties, such as elastic, electronic, and thermodynamic properties are not well established. Here, we conduct first-principles calculations to investigate the fundamental physical properties of this material. The structures are optimized by full relaxation, and the lattice parameters are obtained. The elastic properties (bulk modulus, shear modulus, Young’s modulus, Poisson’s ratio, and anisotropy factor) are calculated and discussed, and the Debye temperatures of three phases are estimated from the calculated elastic constants. The density of states is calculated to study the structural stability mechanism. At the end, we discuss the changes of free energy, entropy, specific heat, and thermal expansion with temperature.
The rest of the present paper is organized as follows. In Section 2, the methods of the computations are described in detail. The results and discussion are given in Section 3. Finally, we draw our conclusions briefly in the last section.
2. Methods of the computations
First-principles calculations in this work are performed in the Vienna ab initio Simulation Package (VASP).[14] We use the generalized gradient approximation with the Perdew–Burke–Ernzerhof (PBE)[15] functional describing the exchange and correlation energy. The projector augmented-waves (PAW) potential[16] is used, of which the valence configuration for Mg is 2p63s2. For Ag and Sb, the valence electron configurations are 4d105s1 and 5s25p3, respectively. Convergences of structure optimization and calculations of physical properties with respect to the plane wave cutoff energy and k-points are tested. For structural optimization, a cutoff energy of 500 eV and k-point of 6×6×6 are used. Atomic and cell variables are simultaneously relaxed until all the residual forces are smaller than 1 meV/Å. For the calculations of electronic structure, elastic constants and vibration properties, a cutoff energy of 500 eV and Monkhorst and Pack[17]k-points of 18×18×18 are used. The density of states is calculated with k-points 20×20×20. The whole set of force constants is obtained from Hellmann–Feynman (HF) forces generated by a nonequivalent atomic displacement in a supercell for a given crystal structure. In the present study, the size of the supercell is 2×2×2 of the primitive cells for three phases. The k-point mesh of 12×12×12 is chosen for this model. A dynamical matrix is constructed from HF forces acting on all atoms in the supercells with a displaced atom. The maximum HF force is within 0.02 eV/Å. Total energies are calculated using a set of supercells with nonequivalent atomic displacements for each polymorph. The number of supercells for the α, β, and γ phases are 42, 21, and 3, respectively. Furthermore, phonon related thermal properties such as the entropy, Gibbs energy and lattice heat capacity are evaluated by employing the quasiharmonic approximation (QHA).[18]
3. Results and discussion
3.1. Structural properties
Structure optimization of the MgAgSb alloy is first performed to determine the lattice constants and the atomic positions of high-temperature γ phase, intermediate-temperature β phase and room temperature α phase, based on the experimental data.[8] The optimized structures are shown in Figs. 1(a)–1(c). The γ phase is a cubic half-Heusler structure with space group where Mg, Ag, and Sb atoms occupy the 4b (0.5,0.5,0.5) site, 4c (0.25,0.25,0.25) site, and 4a (0,0,0) site, respectively. The β MgAgSb structure is tetragonal (P4/nmm), and Mg, Ag, Sb atoms are located at Wyckoff positions 2c (0.25,0.25,0.2957), 2a (0.75,0.25,0), and 2c (0.25,0.25,0.7184), respectively. The α structure is also tetragonal (space group ), in which Mg and Sb atoms are situated at 16i (0.964,0.296,0.096), 16i (0.239,0.475,0.120), respectively. For Ag, three sites 4a (0.0,0.0,0.25), 4b (0.0,0.0,0.0), 8e (0.224,0.224,0.25) are all occupied. In these three structures, the Mg and Sb atoms form a rocksalt-type structure, which is distorted at the intermediate and room temperature. The optimized unit cell parameters and experimental values are listed in Table 1 for comparison. The calculated values of lattice constants are in excellent agreement with the experimental[8] and other theoretical values[19] with differences less than 2%, which demonstrates the effectiveness of the proposed simulation model. The calculated total energies of our considered systems (γ,β,α) are –9.129845, –9.1918745, and ߝ9.197573 eV/f.u. respectively, which indicates that the phase transitions from γ to β, then to α for MgAgSb alloy are in accordance with the experimental results.[8]
Fig. 1. Crystal structures of MgAgSb: (a) γ-phase, (b) β-phase, (c) α-MgAgSb with the Mg shown in turquoise, the Sb in light tan, and the Ag in silver inside the polyhedra.
Table 1.
Table 1.
Table 1.
Lattice constants and atomic positions of MgAgSb.
.
Phase
a
b
c
Atomic position
γ-MgAgSb
6.576
6.576
6.576
Mg (0.5 0.5 0.5), (0.5 0.5 0.5)a, (0.5 0.5 0.5)b
6.564a
6.564a
6.564a
Ag (0.25 0.25 0.25), (0.25 0.25 0.25)a, (0.25 0.25 0.25)b
The elastic properties describe the mechanic properties of materials under the strain, while the strain is a very familiar structural distortion in material fabrication. Thus, they are useful for understanding their responses to pressure, mechanical strength, and phase transition. Elastic constants can be calculated by applying small strains to the equilibrium state and determining the corresponding energy variations. In a cubic lattice, the number of independent elastic constants is three, while the tetragonal lattice has six independent elastic constants. The calculated elastic constants of three structures are listed in Table 2. For cubic γ-MgAgSb, we obtain C11 = 70.6 GPa, C12 = 52.1 GPa, C44 = 29.8 GPa, which fulfil the mechanical stability conditions of cubic system [(C11 + 2C12) > 0, C44 > 0, C11 > 0, (C11 − C12) > 0].[20,21] The obtained elastic constants of tetragonal phases also satisfy the stability conditions:[22] (C11 − C12) > 0, (C11 + C33-2C13) > 0, (2C11 + C33 + 2C12 + 4C13) > 0, C11 > 0, C33 > 0, C44 > 0, C66 > 0. To the best of our knowledge, neither experimental nor theoretical values of elastic moduli for the MgAgSb alloys are available in the literature. Elastic constants can be used to determine the mechanical properties such as bulk modulus (B), shear modulus (G), Young’s modulus (E), Poisson’s ratio (ν), and elastic anisotropic factor (A) by using the Voigt–Reuss–Hill averaging scheme (VHR).[23–25] For the cubic system, the Voigt bounds of B and G are
and the Reuss bounds of B and G are
The bulk modulus and shear modulus are estimated by Hill’s average as follows: B = (BV + BR)/2, and G = (GV + GR)/2. For the tetragonal lattice, the B and G are given by VHR approximation.[24,25] Apart from this, the E, ν, and A are calculated with the method explained in our previous work.[26,27] These properties are also presented in Table 2. A ratio of B/G is usually used to differentiate between the ductile and brittle character of material.[28] The critical value is 1.75. If B/G < 1.75, the material behaves in a brittle manner, otherwise, it is regarded as a ductile material. According to Table 2, it can be seen that the three values are all larger than 1.75, which indicates that the three structures have a ductile feature each. Considering the ductile character of metal, it is not unexpected that MgAgSb is ductile material and is easily processed. With the increase of temperature, the value of E also increases, indicating that the temperature enhances the stiffness of this material. Poisson’s ratios of the three phases are larger than 0.25 and less than 0.5, suggesting that interatomic forces in three structures are central.[29] The calculated elastic anisotropy factor reduces quickly from α to γ, showing that the MgAgSb alloy is more isotropic with temperature increasing, which is in accordance with the experimental result.[8]
Table 2.
Table 2.
Table 2.
Calculated values of elastic constants (Cij in unit GPa), bulk modulus (B in unit GPa), shear modulus (G in unit GPa), and Young’s modulus (E in unit GPa), Poisson’s ratio (ν), elastic anisotropic factor (A), transverse elastic velocity (νt in unit m/s), longitudinal elastic velocity (νl in unit m/s), average elastic velocity (νm in unit m/s), elastic Debye temperature (TD in unit K) and for the three MgAgSb phases at the theoretical equilibrium volume.
Calculated values of elastic constants (Cij in unit GPa), bulk modulus (B in unit GPa), shear modulus (G in unit GPa), and Young’s modulus (E in unit GPa), Poisson’s ratio (ν), elastic anisotropic factor (A), transverse elastic velocity (νt in unit m/s), longitudinal elastic velocity (νl in unit m/s), average elastic velocity (νm in unit m/s), elastic Debye temperature (TD in unit K) and for the three MgAgSb phases at the theoretical equilibrium volume.
.
In a word, calculation results reveal that the MgAgSb has a low value of modulus and a high value of Poisson’s ratio (> 0.25). Further, it is a soft (ductile) material due to strong metallic bonding.
As Debye temperature (TD) is closely related to many physical properties such as specific heat, elastic constants, thermal coefficient and melting temperature, we also calculate the Debye temperature from the following equation:[30]
where h is the Planck constant, k is the Boltzmann constant, NA is the Avogadro number, n is the number of atoms per formula unit, M is the molecular mass per formula unit, and ρ is the density. The average sound velocity νm is given by[30,31]
where νt and νl are the longitudinal and transverse elastic wave velocities of the polycrystal1ine materials, respectively and are obtained by Navier’s equations:[30]
The shear sound velocity vt, longitudinal sound velocity vl, average wave velocity vm, and Debye temperatures TD at 0 K are calculated and listed in Table 2. The TD values of γ-, β-, α-MgAgSb obtained are 280 K, 145 K, 139 K respectively. Li et al.’s results showed that the Debye temperature of a-MgAgSb is 160 K, the longitudinal and transversal phonon velocities 3708 m/s and 1120 m/s, respectively,[32] which are close to our calculated TD, vl, and vmα-MgAgSb. The diminishing TD is connected with the distorted lattice structure and bond strength. Future experiments will testify our calculated results.
3.3. Electronic properties
In order to have further insights into the bonding characteristics of MgAgSb and reveal the underlying mechanism for the mechanical properties and structural stability, we calculate the total density of states (TDOS) and partial density of states (PDOS) of the three phases as shown in Figs. 2(a)–2(c). From Fig. 2(a) to Fig. 2(c), it is found that the TDOS at the Fermi level is 0.4480 states/eV per unit cell for γ-MgAgSb, 0.3891 states/eV per unit cell for the β phase, 0.107 states/eV per unit cell for the α phase, respectively, which is very close to the TDOS values by Miao and Ghosez [19] and Ying et al.[9] Especially for the α phase at low temperature, we also calculate its TDOS with hybrid HSE06 functional and find that the TDOS value has very small difference at Fermi level, which indicates that the α structure is semimetal, which is in accordance with the result in Ref. [19]. The calculated TDOS at the Fermi level indicates that the γ phase is most unstable, and the α phase is most stable, which is consistent with the experimental results.[8] Besides, we find that a pseudogap exists near the Fermi level for low temperature α phase, suggesting that it is the most stable structure. For the PDOSs of γ, β, α in Fig. 2, Ag-4d states and Sb-5p states mainly contribute to the lower part of the valence band, which shows the obvious hybrid character of Ag-4d and Sb-5p states. A hybrid character is simultaneously observed in the conduction band composed mostly of Mg-3s, Ag-4d, and Sb-5p.
Fig. 2. Calculated total and partial densities of states of MgAgSb compound: (a) α-MgAgSb, (b) β-MgAgSb, (c) γ-MgAgSb. The Fermi level is set to 0 eV.
3.4. Thermodynamics properties
Thermal properties play an important role in understanding the thermal response of a solid. The temperature dependence of the thermodynamic property of γ-MgAgSb was evaluated in a quasi-harmonic approximation.[33] The details of these calculations have been provided previously.[34,35] The calculated free energy, entropy, and specific heat (Cv) of high temperature γ-MgAgSb are shown in Fig. 3(a). It is known that the free energy of a structure is closely related to its geometrical structure. From the free energy curve shown in Fig. 3(a), it is identified that the free energy decreases as temperature increases. Figure 3(a) shows that as the temperature increases the entropy curves increase smoothly as well. This behavior is understandable because the phonon frequency should increase with temperature. It is observed that in a temperature range of 0 K–1000 K the calculated Cv increases sharply with increasing temperature while at low temperatures, then increases slowly with further increasing temperature. The obtained Cv curve in a range of 0 K–300 K is close to the low temperature Cv measurement by Li et al.,[32] which indicates that our calculations are reasonable. At high temperatures, the Cv is close to the classical Dulong–Petit limit (74.8 J/mol·K). This indicates that the optical and acoustic modes are all excited at the temperature. Furthermore, the variation of thermal expansion coefficient α with temperature is illustrated in Fig. 3(b). At zero pressure the α increases exponentially at low temperature and gradually approaches a linear increase for T > 150 K. Also, the α value varies from 70×10−6/K to 100×10−6/K at temperatures in the range 300 K–700 K, which is slightly more than the experimental value 64×10−6/K at temperatures in the range 300 K–693 K.[8]
Fig. 3. (a) The calculated thermodynamic properties in the temperature range 0 K–1000 K: vibrational Helmholtz free energy (red curves), vibrational entropy (blue curves), and specific heat at constant volume Cv (gree curves). (b) The calculated thermal expansion (α) of high temperature in a temperature range of 0 K–800 K.
4. Conclusions
In this work, an attempt is made to employ the first-principles method to investigate the structural, elastic, electronic, and thermodynamic properties of the MgAgSb material. The calculated lattice parameters of three MgAgSb structures are in good agreement with the available experimental data. The Cij, B, G, E, ν, and A are determined, showing that the three MgAgSb structures with ductile feature are mechanically stable. The density of states is analyzed and shows that the α-MgAgSb is most stable with a pseudogap near the Fermi level. Finally, the thermodynamic properties are also studied. The thermodynamic properties such as free energy, entropy, Cv, and α are predicted theoretically, demonstrating that the results from the first-principles calculations are useful. At 0 GPa and 300 K, the heat capacity Cv is 74.8 J mol−1·K−1 and the thermal expansion coefficient α is 5.19×10−5 K−1. As there are few experimental data at present, it is necessary to compare these results quantitatively with reliable experimental data in the future, to discuss the calculation accuracy in detail. In addition, the results calculated in this work provide the theoretical reference for experimentally designing the thermoelectric materials at even higher temperature.






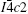








{kind=link}
{kind=link}
{kind=link}
 , Wang Jun-Tao1, Li Xiao-Dong1, Jiang Zhen-Yi2]
, Wang Jun-Tao1, Li Xiao-Dong1, Jiang Zhen-Yi2]