First-principles calculation of the structural, electronic, elastic, and optical properties of sulfur-doping
Huang Chang-Bao, Wu Hai-Xin†,  , Ni You-Bao, Wang Zhen-You, Qi Ming, Zhang Chun-Li
, Ni You-Bao, Wang Zhen-You, Qi Ming, Zhang Chun-Li
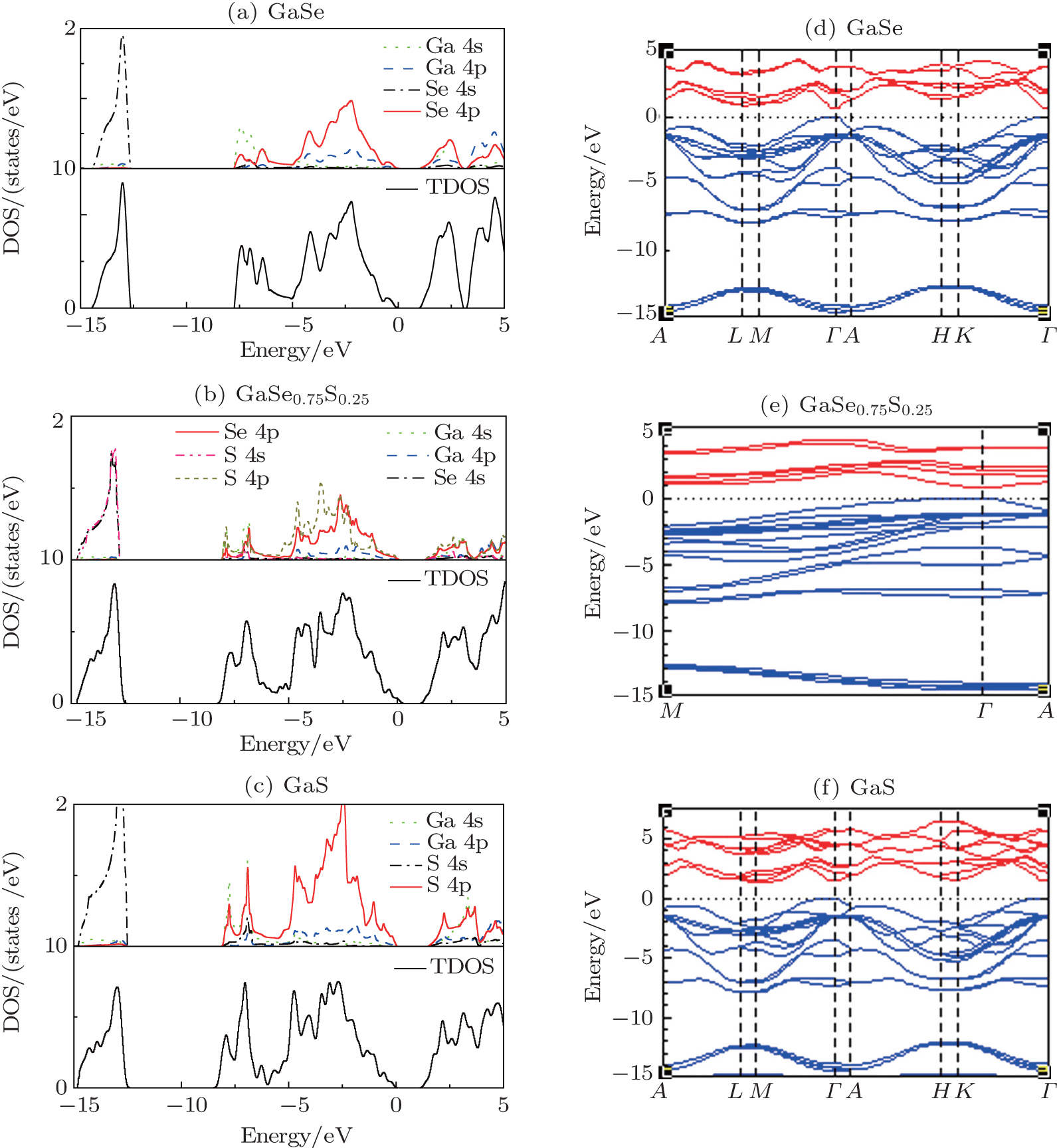
, Ni You-Bao, Wang Zhen-You, Qi Ming, Zhang Chun-Li Total and partial density of states (left) and electronic band structure (right) of