




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Band structure, Fermi surface, elastic, thermodynamic, and optical properties of AlZr3 , AlCu3 , and AlCu2 Zr: First-principles study
[Parvin R1, Parvin F1, Ali M S2, †,  , Islam A K M A3]
, Islam A K M A3]
, Islam A K M A3]
|
|


† Corresponding author. E-mail:
The electronic properties (Fermi surface, band structure, and density of states (DOS)) of Al-based alloys AlM3 (M = Zr and Cu) and AlCu2Zr are investigated using the first-principles pseudopotential plane wave method within the generalized gradient approximation (GGA). The structural parameters and elastic constants are evaluated and compared with other available data. Also, the pressure dependences of mechanical properties of the compounds are studied. The temperature dependence of adiabatic bulk modulus, Debye temperature, specific heat, thermal expansion coefficient, entropy, and internal energy are all obtained for the first time through quasi-harmonic Debye model with phononic effects for T = 0 K–100 K. The parameters of optical properties (dielectric functions, refractive index, extinction coefficient, absorption spectrum, conductivity, energy-loss spectrum, and reflectivity) of the compounds are calculated and discussed for the first time. The reflectivities of the materials are quite high in the IR–visible–UV region up to ∼ 15 eV, showing that they promise to be good coating materials to avoid solar heating. Some of the properties are also compared with those of the Al-based Ni3Al compound.
Aluminum-based alloys have received much attention in recent years because of their important roles in many technological applications. They have vast applications in the aerospace, microelectronic, motorized vehicles, and domestic industry. As the alloys are important in the engineering and research areas, mechanical behavior has become a focus of various investigations. A large quantity of alloy elements can be used to form the Al-based intermetallic compounds. It is observed that the bonding behaviors and electronic natures are changed of such a type of alloys which show excellent mechanical, physical, and chemical properties.[1] The compounds involving aluminum and transition metals are known to have high resistance to oxidation and corrosion, elevated-temperature strength, relatively low density, and high melting points, which make them desirable candidates for high-temperature structural applications.[2] Zirconium can effectively enhance the mechanical strengths of the alloys when copper and zinc elements exist in aluminum and Al-based alloys.[3] Many investigations have focused on the constituent binary systems, such as Al–Cu, Al–Zr, and Cu–Zr.[4–9] Some theoretical investigations have been performed on Al-based alloys such as structural, elastic and electronic properties.[10,11]
To the best of our knowledge, electronic band structures, Fermi surfaces, thermal and optical properties of AlZr3, AlCu3, and AlCu2Zr compounds have not yet been discussed theoretically and experimentally. The thermodynamic properties include a variety of properties and phenomena such as bulk modulus, specific heat, thermal expansion coefficient, Debye temperature, etc. The specific heat of a material is one of the most important thermodynamic properties indicating its heat retention or loss ability. We have also briefly discussed our calculated structural and elastic properties for AlZr3, AlCu3, and AlCu2Zr intermetallic alloys.
On the other hand, the optical properties of solids provide an important tool for studying energy band structures, impurity levels, localized defects, lattice vibrations, and certain magnetic excitations. The optical conductivity or the dielectric function indicates a response of a system of electrons to an applied field. Thus there is a need to deal with all these issues which have been covered for the first time in the present paper. A detailed discussion and analysis are made, some of which are in comparison with the results of available Al-based alloys.[10,12]
The density functional theory (DFT) based on the CASTEP code has been used in the present investigation.[13–16] The interactions between valence electrons and ions are considered using the ultrasoft pseudo-potential formalism of Vanderbilt.[17] To find out the minimum potential energy of the compound, a quasi-Newton (variable-metric) minimization method, the Broyden–Fletcher–Goldfarb–Shanno (BFGS) update scheme,[18] is utilized, which provides a very efficient and fast way to explore the optimizing crystal structure. The electron wave function is expanded in plane waves up to an energy cutoff of 400 eV for AlZr3 and 500 eV for AlCu3 as well as AlCu2Zr for the sampling of the Brillouin zone,[19] a 15×15×15, 21×21×21, 9×9×9 Monkhorst–Pack mesh are employed[20] for AlZr3, AlCu3, and AlCu2Zr, respectively. Generalized gradient approximation (GGA) of the Perdew–Burke–Ernzerhof for solids (PBEsol) formalism,[16] which is dependent on both the electron density and its gradient at each space point is used to evaluate the exchange-correlation energy. Optimization is performed using a convergence threshold of 10−5 eV/atom for the total energy and 10−3 Å for maximum displacement. Maximum force and stress are 0.03 eV/Å and 0.05 GPa, respectively for all the calculations. Norm conserving pseudo-potential is used only for calculating the optical properties, keeping the other setup unchanged. The resulting stress can be calculated with respect to the optimized crystal structure[21] using a set of homogeneous deformations with a finite value.
The thermodynamic properties are studied within the quasi-harmonic Debye model implemented in the Gibbs program,[22] a detailed description of which can be found in Refs. [23]– [25]. We use the DFT calculated E ∼ V (energy ∼ volume) data at T = 0 K, P = 0 GPa, and the third-order Birch–Murnaghan EOS.[26] Using this, the thermodynamic properties such as the bulk modulus, thermal expansion co-efficient, specific heats, and Debye temperature at non-zero temperature and pressure are calculated.
Aluminum-based intermetallic AlM3 (M = Zr and Cu) binary alloys crystallize into the cubic Cu3Au structure[27,28] with space group Pm-3m (No. 221) consisting of Al atoms at the corners and M atoms at the face centers of the cube. The atomic positions are Al: 1a (0,0,0), and M: 3c (0,1/2,1/2). The ternary AlCu2Zr alloy is a partially ordered Cu2MnAl-type fcc (space group No. 225: Fm-3m) structure,[29] where Al atoms occupy the 4a Wyckoff site (0,0,0), Cu atoms occupy the 8c site (1/4,1/4,1/4) and Zr atoms occupy the 4b site (1/2,1/2,1/2). In order to investigate the ground-state properties of AlM3 (M = Zr and Cu) and AlCu2Zr alloys, the geometry optimizations are performed first with the full relaxation of the cell shape and atomic positions. The equilibrium crystal structures of these compounds are shown in Fig.
 | Fig. 1. Crystal structures of AlM3 (M = Zr and Cu) and AlCu2Zr. |
| Table 1. Values of calculated and experimental lattice constant a (in unit Å), equilibrium volume V0 (in units Å3), bulk modulus B0 (in unit GPa) of AlZr3, AlCu3, and AlCu2Zr alloys. . |
Generally, elastic constants of a solid permit us to obtain the mechanical properties and they can be used to describe the resistance of a crystal to an externally applied stress. They also provide important information about bonding characteristics near the equilibrium state. Thus, it is essential to investigate the elastic constants to understand the mechanical behavior of AlM3 (M = Zr and Cu) and AlCu2Zr compounds at equilibrium and under pressure. For a material with cubic symmetry, there are only three independent elastic constants, i.e., C11, C12, and C44. In this paper, the calculated single crystal elastic constants of these compounds are listed in Table
| Table 2. Values of calculated elastic constants Cij (in unit GPa), bulk modulus B (in unit GPa), shear modulus G (in unit GPa), Young’s modulus E (in unit GPa), Poisson’s ratio ν, elastic anisotropic factor A for AlZr3, AlCu3, and AlCu2Zr alloys at equilibrium. . |
The pressure dependences of the calculated elastic constants of AlZr3, AlCu3, and AlCu2Zr are plotted in Fig.
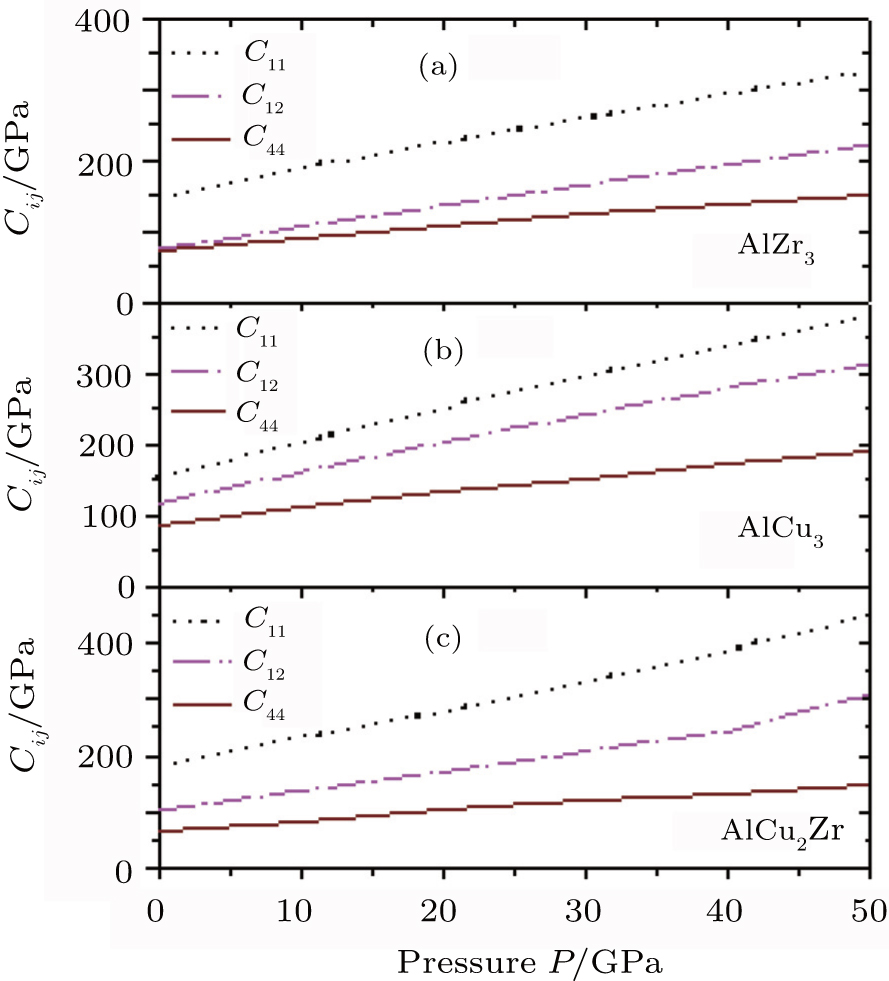 | Fig. 2. Pressure dependences of elastic constants of (a) AlZr3, (b) AlCu3, and (c) AlCu2Zr. |
The polycrystalline elastic properties, such as bulk modulus B, shear modulus G, Young’s modulus E, and Poisson’s ratio ν are estimated from the calculated single crystal elastic constants, and are presented in Table
The pressure effects on the calculated bulk modulus, shear modulus, and Young’s modulus of AlZr3, AlCu3, and AlCu2Zr are shown in Fig.
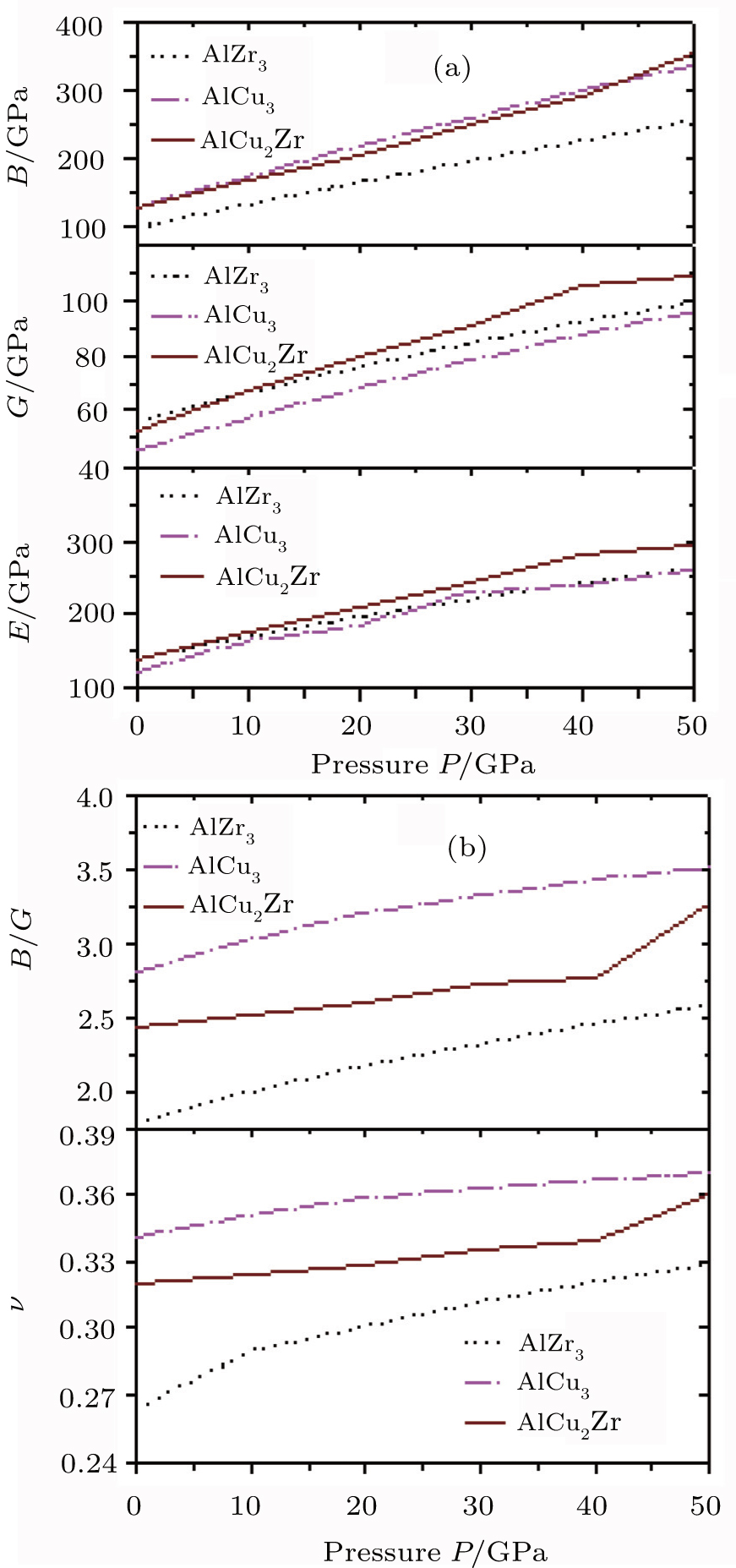 | Fig. 3. Pressure effects on (a) bulk modulus (B), shear modulus (G), and Young’s modulus (E); (b) the values of ratio of bulk modulus to shear modulus (B/G) and Poisson’s ratio (ν) of AlZr3, AlCu3, and AlCu2Zr. |
Figure
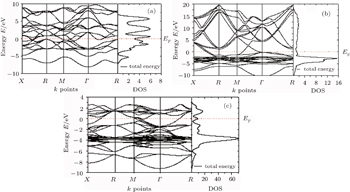
The investigated electronic band structures along high symmetry directions in the Brillouin zones together with the total densities of states (DOSs) of AlZr3, AlCu3, and AlCu2Zr are depicted in Fig.
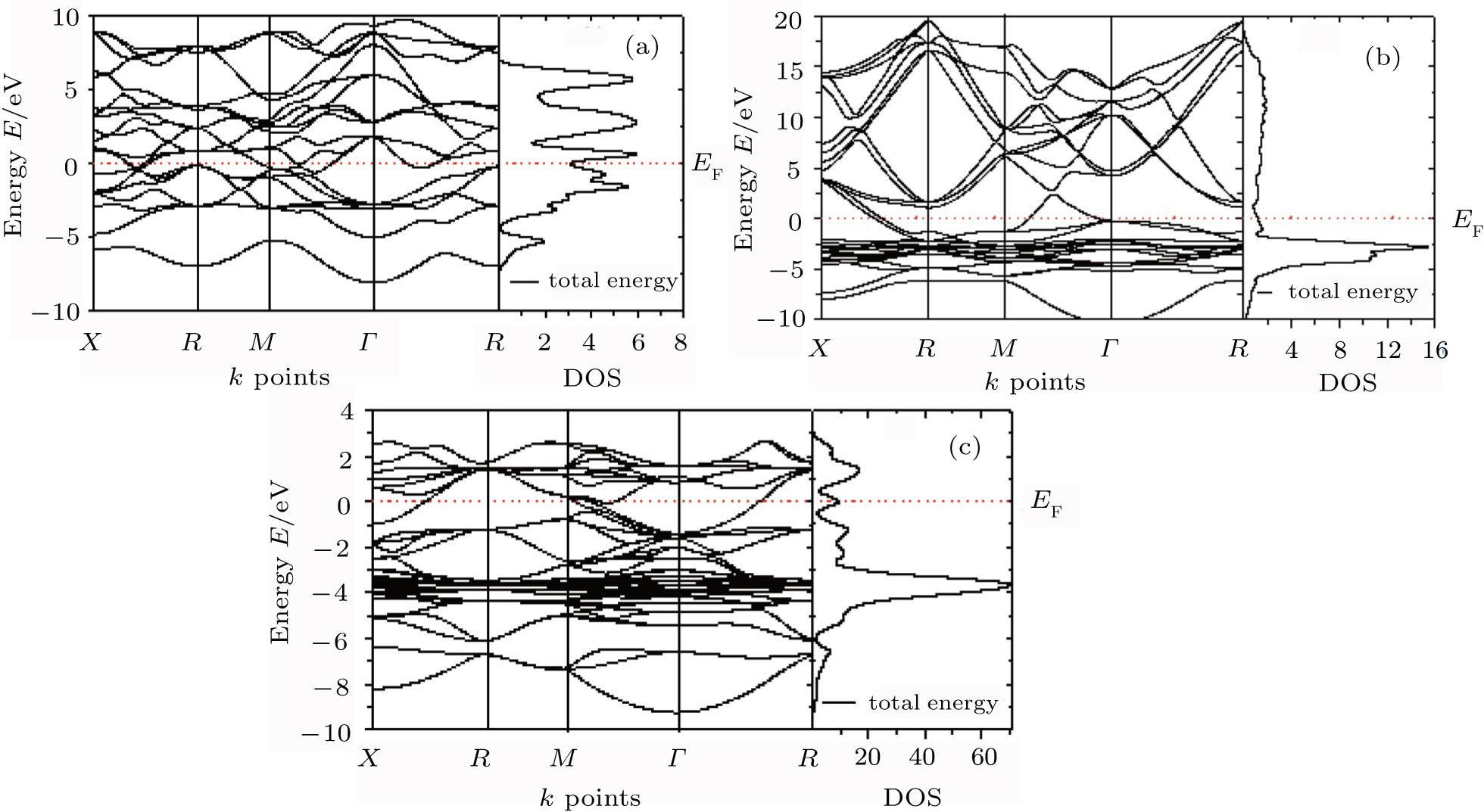 | Fig. 4. Band structures and total DOSs of (a) AlZr3, (b) AlCu3, and (c) AlCu2Zr. |
It is observed from Fig.
According to the covalent approach, the guiding principle is to maximize bonding. Hence the stability increases with increasing occupancy in the bonding region, for a series of compounds having the same structure.[35] Therefore it can be said that AlZr3 is structurally more stable than AlCu3. On the other hand, for AlCu2Zr, the main bonding peaks from Fermi level to 3 eV predominantly derive from the Zr 4d orbitals. The phase stability of intermetallic compound depends on the location of the Fermi level and the value of the DOS at EF.[36,37] The lower value of DOS at EF corresponds to a more stable structure. The values of the total DOS at the Fermi level in states are 3.2 eV and 9.3 eV for AlZr3 and AlCu2Zr alloys, respectively. Therefore, it can be said that AlZr3 has a more stable structure than the other two alloys of Al under consideration, which agrees very well with the observation in Ref. [10].
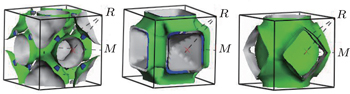
The shape of the Fermi surface is derived from the periodicity and symmetry of the crystalline lattice and from the occupation of electronic energy bands. It separates unfilled orbitals from the filled orbitals. The Fermi surface topologies of AlZr3, AlCu3, and AlCu2Zr are shown in Fig.
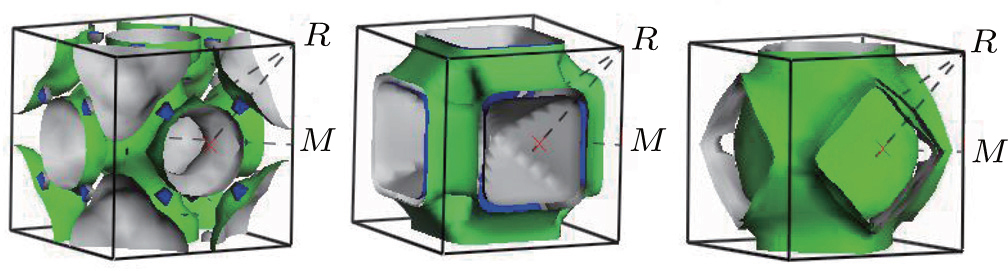 | Fig. 5. Fermi surfaces of (a) AlZr3, (b) AlCu3, and (c) AlCu2Zr. |
Charge transport is mainly limited by carrier–carrier and carrier–phonon scattering. Besides, there is always impurity scattering in most real systems. The carrier–carrier scattering is intimately related to the electronic band structure and density of states at the Fermi level. The topology of the Fermi surface also plays a role. For example, the highly dispersive nature of energy dispersion curve for AlZr3 (Fig.
The thermodynamic properties such as the bulk moduli, specific heats, Debye temperatures, and volume thermal expansion coefficients at different temperatures and pressures are evaluated for the first time for these compounds. We calculate all the thermodynamic properties in a temperature range from 0 K to 1000 K and a pressure range from 0 GPa to 50 GPa, where the quasi-harmonic Debye model remains valid.
In the quasi-harmonic model, the nonequilibrium Gibbs function G∗(V;P,T) can be written as[22]


The temperature dependences of adiabatic bulk modulus, B of AlZr3, AlCu3, and AlCu2Zr alloys at P = 0 GPa are shown in Fig.
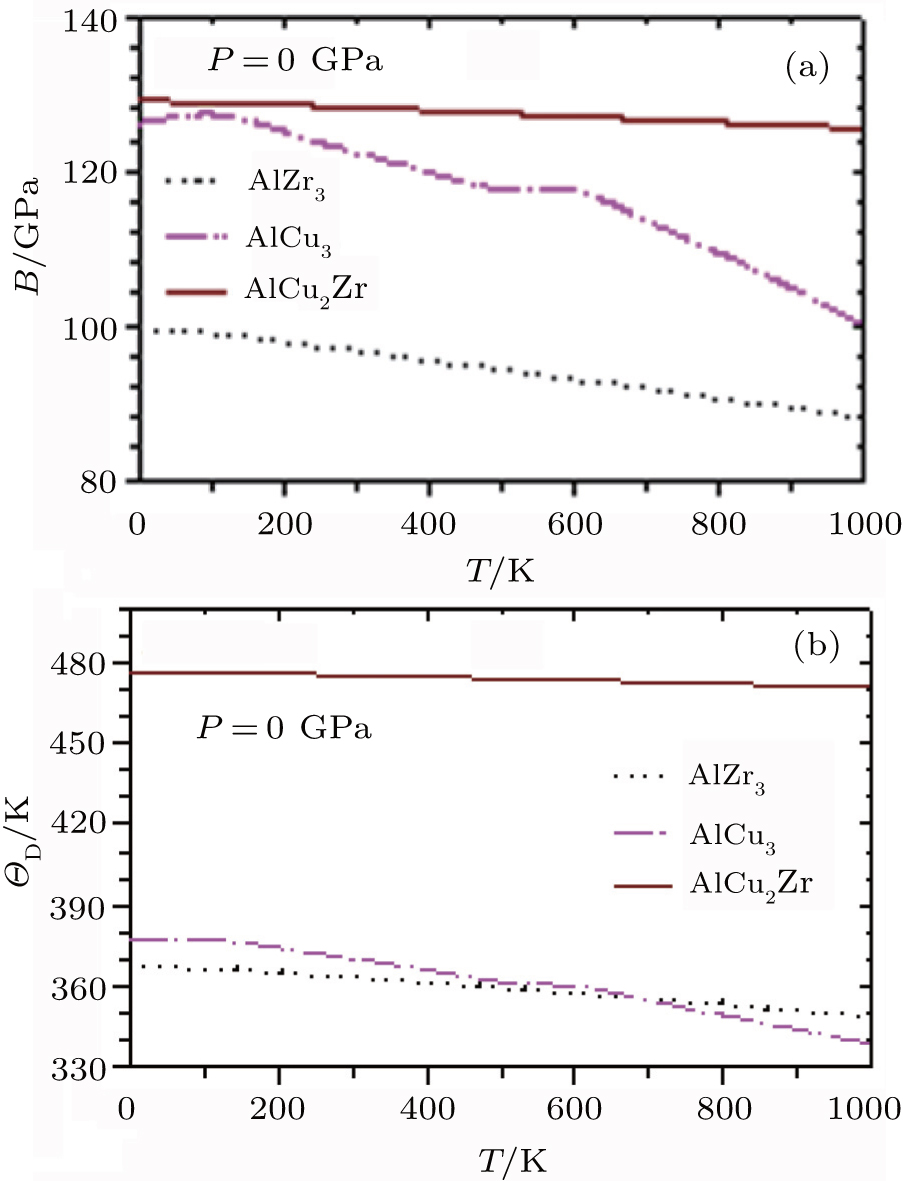 | Fig. 6. Temperature dependences of (a) adiabatic bulk modulus and (b) Debye temperature of AlZr3, AlCu3, and AlCu2Zr. |
Debye temperature is basically a measure of the vibrational response of the material. Debye temperature is not a strictly determined parameter; various estimates may be obtained through well-established empirical or semi-empirical formula, relating ΘD to various macroscopic properties.[39] Figure
It is observed from Fig.
The specific heat of the material is usually approximated by the contribution of the lattice specific heat. Figure 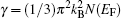
 | Fig. 7. Specific heats at constant (a) pressure and (b) volume versus temperature of AlZr3, AlCu3, and AlCu2Zr. |
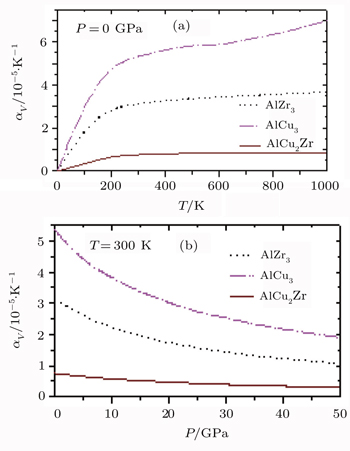
Finally, the computed results on the variations of the volume thermal expansion coefficient, αv with temperature and pressure for AlZr3, AlCu3, and AlCu2Zr alloys are displayed in Fig.
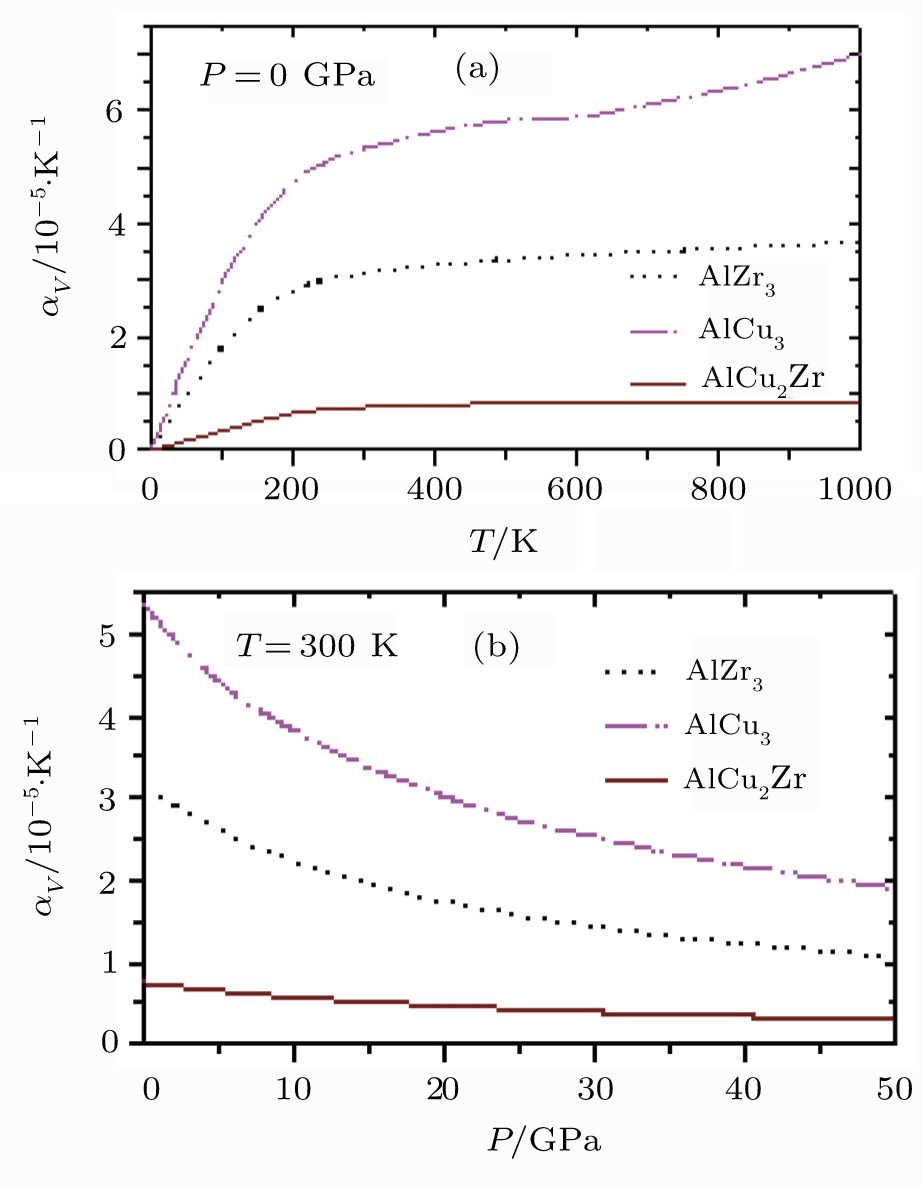 | Fig. 8. Variations of αv with (a) temperature and (b) pressure of AlZr3, AlCu3, and AlCu2Zr. |
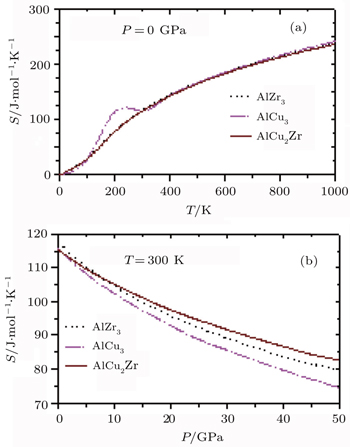
The variations of entropy, S of AlZr3, AlCu3, and AlCu2Zr alloys each as a function of temperature and pressure are shown in Fig.
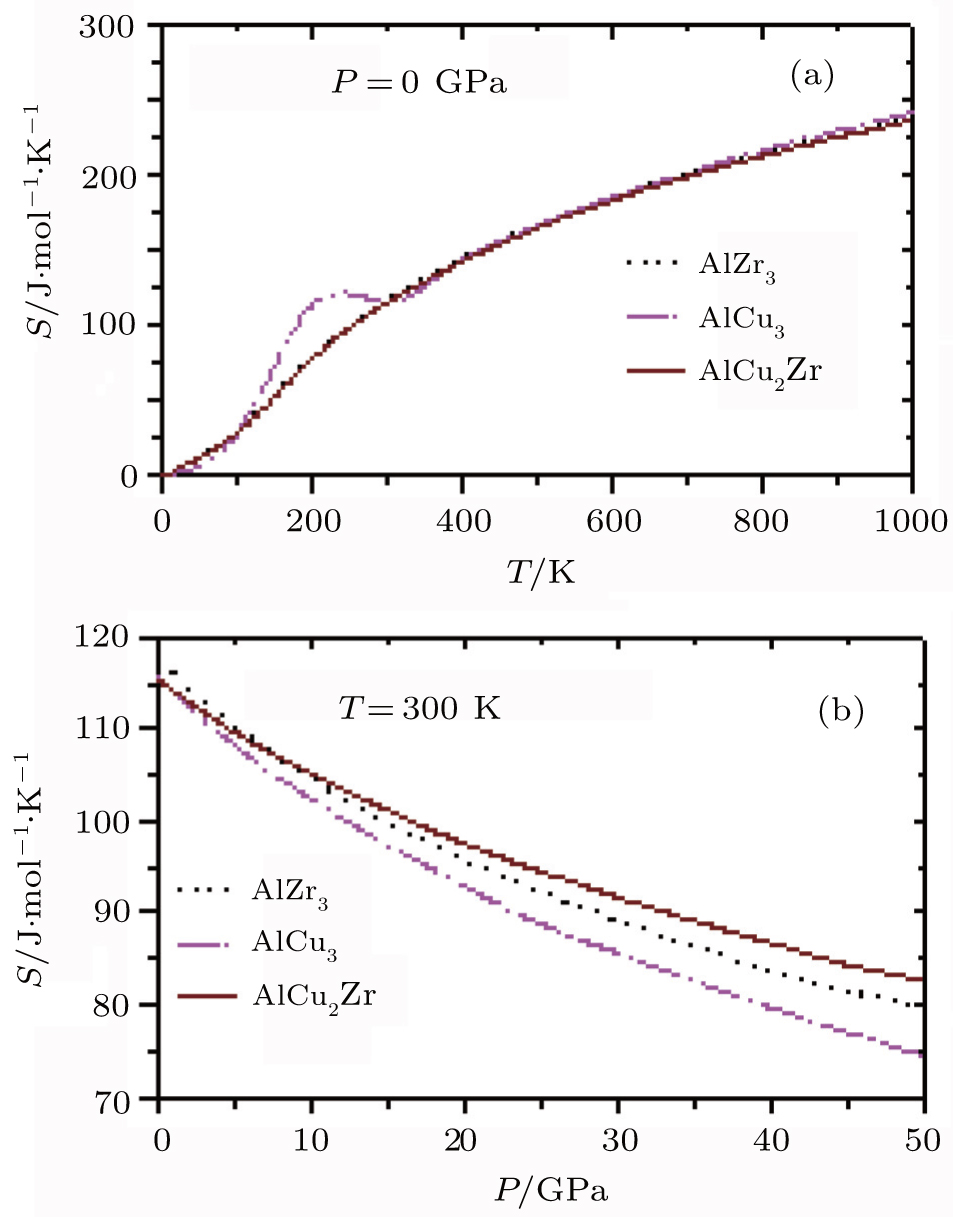 | Fig. 9. Variations of S with (a) temperature and (b) pressure for AlZr3, AlCu3, and AlCu2Zr. |
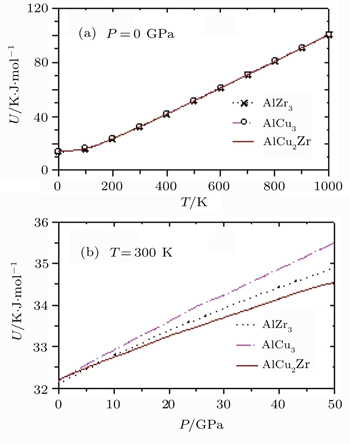
The temperature and pressure variations of the calculated internal energies for AlZr3, AlCu3, and AlCu2Zr alloys are shown in Fig.
 | Fig. 10. (a) Temperature and (b) pressure dependences of U for AlZr3, AlCu3, and AlCu2Zr. |
The optical properties of AlZr3, AlCu3, and AlCu2Zr alloys may be derived from the knowledge of the complex dielectric function, ɛ(ω). Generally, there are two contributions for ɛ(ω), namely from intraband and interband transitions. The contribution from the latter is vital only for metals.[41] The complex dielectric function can be expressed as ɛ(ω) = ɛ1(ω)+ iɛ2(ω). The imaginary part ɛ2(ω) is obtained from the momentum matrix elements between the occupied and unoccupied wave functions within the selection rules, whereas the real part ɛ1(ω) of dielectric function can be derived from the imaginary part using the Kramers–Kronig relation. The knowledge of both ɛ1(ω) and ɛ2(ω) allows the calculation of important optical constants such as the refractive index n(ω), extinction coefficient k(ω), conductivity, optical reflectivity R(ω), absorption coefficient α(ω), and energy-loss spectrum L(ω) using the expressions given in Ref. [42]. The calculational approaches are well established and widely available in the literature[43,44] and hence will not be repeated here.
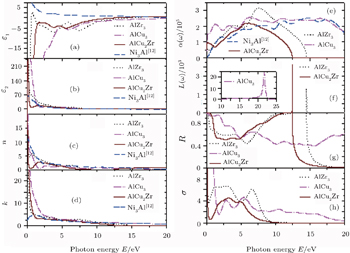
The estimated optical properties of AlZr3, AlCu3, and AlCu2Zr alloys at ground state are presented in Fig.
 | Fig. 11. (a) Real and (b) imaginary parts of dielectric function, (c) real and (d) imaginary parts of refractive index, (e) absorption, (f) loss function, (g) reflectivity, and (h) real part of conductivity of AlZr3, AlCu3, and AlCu2Zr. |
The plasma frequencies of 10 eV, 4 eV, 6 eV, and damping values of 0.07 eV, 0.05 eV, 0.02 eV are used for AlZr3, AlCu3, and AlCu2Zr alloys, respectively. For all calculations, we use a 0.5-eV Gaussian smearing. The real part ɛ1 goes through zero from below about 14.52 eV, 21.66 eV, 12.47 eV and the imaginary part ɛ2 approaches zero damping values of 14.98 eV, 22.80 eV, and 12.98 eV for AlZr3, AlCu3, and AlCu2Zr alloys, respectively. Metallic reflectance characteristics are exhibited in the range ɛ1 < 0.
The velocity of propagation of an electromagnetic wave through a solid is given by the frequency dependent complex refractive index N = n + ik, where the real part n is related to the velocity and the imaginary part k, the extinction coefficient is related to the decay or damping of the oscillation amplitude of the incident electric field. The refractive indexes and extinction coefficients are illustrated in Figs.
Figure
Loss function is intimately related to absorption and reflection. Loss function refers to the fast electron traversing in a material. It is clear from the study of the figure that maximum reflectivity occurs where the minimum absorption takes place. So the study of loss function is the most important in material study. The electron energy loss function L(ω) is depicted in Fig.
The variations of calculated optical reflectivity R(ω) with incident photon energy are displayed in Fig.
For the three alloys, their photoconductivities start with zero photon energy (Fig.
First-principles calculations based on DFT and the quasi-harmonic Debye model are used to investigate different properties of AlZr3, AlCu3, and AlCu2Zr alloys. Based on the calculated results, we find that AlZr3 phase has the highest stiffness among the three phases. On the other hand, the Poisson’s ratio changes from 0.264 to 0.341, which indicates that AlM3 (M = Zr and Cu) and AlCu2Zr compounds show metallic behaviors. In view of Pugh’s criterion for these three alloys it is observed that these three compounds show ductility behavior. Under the effect of pressure, the value of C11 increases rapidly with pressure compared to C12 and C44 for all compounds, which indicates that the longitudinal strain produces a significant change in volume. The larger Poisson’s ratio of AlCu3 compared to those of the other two phases indicates better plasticity.
It is also observed from the DOS value that AlZr3 has a more stable structure than the other two alloys. The pseudogap around the Fermi level indicates the presence of the directional covalent bonding. The AlCu3 and AlZr3 show hole-like Fermi surface whereas AlCu2Zr shows both electron-like and hole-like Fermi surface. The stronger the inter-atomic bonding, the smaller the thermal expansion is. The smaller value of AlCu2Zr indicates stronger atomic bonding whereas the higher value of AlCu3 indicates that it is softer than the other two compounds. The reflectivities of these compounds are high (∼100%) in the IR as well as in the UV region. In the visible region, all the compounds under study may be considered as potential coating materials to avoid solar heating.
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 | |
| 22 | |
| 23 | |
| 24 | |
| 25 | |
| 26 | |
| 27 | |
| 28 | |
| 29 | |
| 30 | |
| 31 | |
| 32 | |
| 33 | |
| 34 | |
| 35 | |
| 36 | |
| 37 | |
| 38 | |
| 39 | |
| 40 | |
| 41 | |
| 42 | |
| 43 | |
| 44 | |
| 45 | |
| 46 | |
| 47 | |
| 48 |