1. IntroductionZn2GeO4 (ZGO) is an important ternary oxide. Recently, ZGO nanomaterial as a kind of nano-luminescence material has become the focus of research, since it has many miraculous new optical properties. The ZGO material possesses fluorescence and wide bandgap properties which are applied to current thin-film photoluminescence devices and field-emission displays, and it is about 40% brighter than the traditional ZnO phosphor.[1–3] In addition, the ZGO can be used as a promising high-capacity anode for lithium batteries due to the excellent rate capability,[4] and as a stable photocatalyst to decompose water and organic pollutant.[5,6] On the other hand, the ZGO was studied as the analog of Mg2SiO4 which is a dominant mineral in the upper mantle initially.[7] Because the Zn2+ with the unfulfilled 3d orbitals displays similarities to Mg2+ and the pressure of Mg2SiO4 phase transition is relatively high, it can be reduced by Ge4+ instead of Si4+, Zn2+ instead of Mg2+. Studying the influence of pressure and temperature on the structural stability and possible phase transition will provide us an insight into the potential applications to extreme conditions.
In this article, we investigate the hexagonal phenacite structure (willemite) of ZGO up to 24 GPa by means of the in-situ x-ray diffraction, high-pressure Raman spectroscopy, and high-pressure and high-temperature (HP-HT) sintering at a cubic press. The pressure-induced amorphization (PIA) is observed at ambient temperature, while the phase transformations from hexagonal phenacite to tetragonal spinel at 1200–1400 °C, 5 GPa, to decomposition above 1500 °C are found. The results are different from some early reports. The ambient-pressure phase of ZGO transformed to cubic spinel and tetragonal spinel at pressures of 3–7.2 GPa,[8] decomposed different minerals at 25–27 GPa, 1000–1400 °C.[12] The composition, pressure, and temperature dependence of the structure transformation are important tools for studying the properties of materials. In general, pressure and temperature can result in disorder in lattice, so we infer that the kinetic hindrance may be one of the mechanisms of PIA. Furthermore, the high-pressure Raman spectrum measurement is firstly performed to illustrate the route of PIA, and the instability of ZGO mainly comes from tilting Zn–O–Ge and Ge–O–Ge bond angles with increasing pressure.
The study aims to better understand the mechanism of PIA, synthesize, and determine the crystal structures of ZGO at HP-HT. There are a few works on the pressure and temperature effect on the properties of materials by HP-HT sintering at a cubic press. The approach particularly suits to obtain and observe the stability of cubic and tetragonal spinel,[8] or investigate these structures of decomposition products from hexagonal phenacite ZGO.[12] Besides, amorphous ZGO possesses better characteristics as anode materials for Li-ion batteries[9] and we can obtain different structures and amorphous bulk solids of ZGO by changing the pressure and temperature at the cubic press.
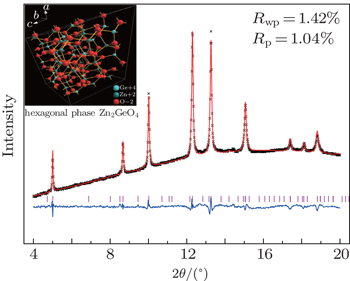
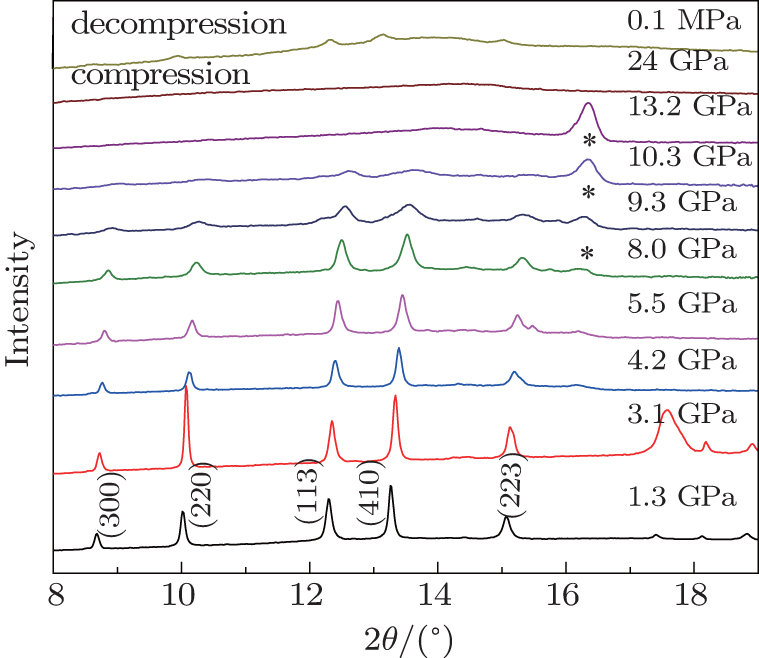
2. Experiment details2.1. Sample preparationZGO was synthesized by solid-state reactions using the starting materials GeO2 (99.9999%) and ZnO (99.9%) in a stoichiometric molar ratio: ZnO:GeO2 = 2:1. The mixed powder was heated to 1200 °C for 12 h in a muffle furnace with a heating rate of 10 °C/min. The annealed sample was confirmed to be the pure hexagonal structure by a powder x-ray diffraction with Cu-Kα radiation at ambient condition, which agrees with the refined result of 1.3 GPa diffraction data shown in Fig. 1. The calculated outline matches the experimental data greatly (RP = 1.02%). The lattice parameters (a = 14.201 Å, c = 9.507 Å) are similar to the result (a = 14.195 Å, c = 9.505 Å) calculated by the GSAS software.
2.2. High-pressure synchrotron radiation angle-dispersive x-ray diffractionThe in-situ high-pressure angle-dispersive x-ray diffraction (ADXRD) measurement was performed at the 4W2 beamline of the Beijing Synchrotron Radiation Facility (BSRF) with a wavelength of 0.6199 Å. The high pressure was generated in a Mao-Bell type diamond anvil cell (DAC) with a pair of 300 μm culet sized diamond anvils. Before the experiment, the ZGO powders and a few ruby grains with a diameter of about 2 μm were loaded into an approximately 100 μm diameter hole drilled in a stainless-steel (T301) gasket and a 4:1 methanol–ethanol was used as a pressure-transmitting medium to decrease the uniaxial stress effect. Then, the gasket was placed on the diamond anvil and compressed to the target pressure at room temperature. Pressure was determined from the Ruby fluorescence shifts and the error is not over 1 GPa.[10] The incident x-ray beam was produced from a Si (111) monochromator and was focused down to 21 μm (vertical) × 8 μm (horizontal) full width at half maximum (FWHM) by a pair of Kirkpatrick–Baez mirrors. The diffraction patterns at various pressures were collected with a Mar345 image plate. The geometrical parameters of the detector were calibrated with a high purity CeO2 powder. An FIT2D software was used to integrate and correct the collected 2D images into one-dimensional profiles. Finally, the diffraction patterns were indexed and refined with the GSAS program packages.
2.3. High-pressure Raman spectrum and HP-HT sinteringThe Raman spectrum measurement was also performed at different pressures using a solid-state laser with a wavelength of 532 nm (RGB laser system, Germany). Prior to the experiment, the Raman system was calibrated using the peak at 520 cm−1 of monocrystalline silicon and the uncertainty is 0.5 cm−1. The high-pressure device was the same as ADXRD’s. The Raman laser beam was focused within ∼5 μm on the surface of the sample, and its power was 50 mW and the exposure time was 10 s.
The HP-HT sintering experiments were performed on a 6 × 14 MN cubic press. During the experiment, the samples were firstly compressed to a target pressure (4–5 GPa) at room temperature, then heated to the temperature (900–1500 °C) for 15 min. Finally, these sintered samples ground to powder were identified and characterized by powder x-ray diffraction at room temperature.
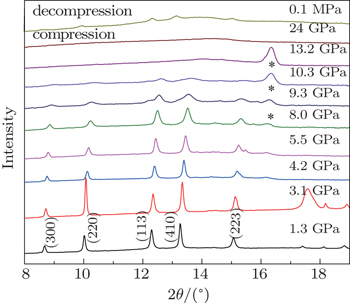
3. Results and discussion3.1. The behaviors of ZGO under HP-HTThe in-situ x-ray diffraction data of ZGO is shown in Fig. 2 for both compression and decompression procedures. The highest pressure applied is 24 GPa. The ADXRD patterns of ZGO indicate that the hexagonal structure remains 8 GPa, but all peaks shift to higher angles, which suggests a pressure-induced lattice contraction. As the pressure increased to 9.3 GPa, the peaks of hexagonal structure were seriously broadened. Above 10.3 GPa, all diffraction peaks disappeared, and the crystalline structure of the sample completely transformed into an amorphous state. The peaks marked by an asterisk may be due to the appearance of the (T301) gasket after amorphization until higher pressure. The amorphization is irreversible, which is evidenced by the pattern of decompression to ambient pressure, but slightly distorted hexagonal structure still remains, and the phenomenon can be interpreted by the “memory” effect of the initial crystal orientation before PIA.[13] It is possible that a local initial structure still exists in a very small domain, the micro-domain is not large enough to x-ray the wave length so that the crystalline phase cannot be displayed.[11] The PIA was confirmed by the high-pressure Raman spectroscopy measurement as well. The pressure at which amorphization starts is 2 GPa lower than x-ray diffraction.
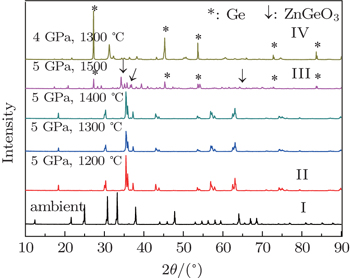
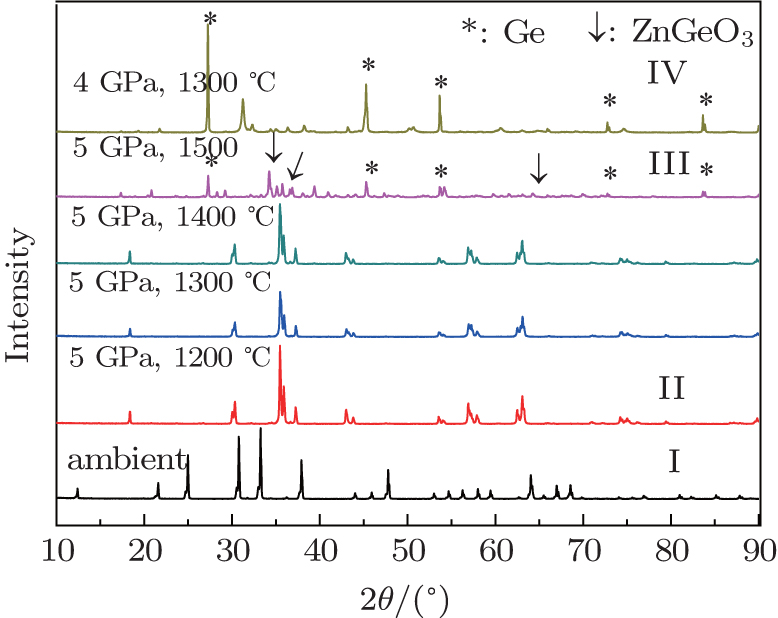
The datum of HP-HT sintering experiments are shown in Fig. 3. Four polymorphs have been identified, and they are designated as phase I–IV in the order of temperature. ZGO undergoes the phase transitions from phase I (willemite) to phase II (tetragonal spinel) at 5 GPa and 1200 °C, which was identified by XRD of the recovered product. Up to 1500 °C, it is possible that a decomposition reaction occurs to the sample of ZGO, we can identify that the decomposition products include Ge and ZnGeO3, the peaks marked by an asterisk and arrows in Fig. 3 indicate cubic phase elemental crystal Ge and hexagonal structure ZnGeO3, respectively, and ZnGeO3 belongs to space groups R-3c (167), but we could not determine other decomposition products and their structure so far. As the temperature reached 1300 °C and the pressure dropped to 4 GPa, the sample may be decomposed into Ge and other oxides. The peaks marked by an asterisk in Fig. 3 have been indexed to cubic phase elemental crystal Ge, which belongs to space groups Fd3m (227) and is different from the condition of 5 GPa, 1300 °C. The structures of phases I–IV are stable at ambient pressure upon the XRD patterns decompressed to ambient pressure. In the early days, some HP-HT works of ZGO were performed in a laser-heated diamond anvil cell. A work on the HP-HT transformation of ZGO has reported the ambient-pressure phase transformed to cubic spinel and tetragonal spinel at pressures of 3 to 7.2 GPa.[8] Upon further compression, Liu reported that the phenakite form of ZGO transformed to a mixture of rock salt phase of ZnO, orthorhombic phase of ZnGeO3 and little distorted spinel phase of ZGO at 25 GPa and 1000–1400 °C, up to 27 GPa, transformed to ZnO, ZnGeO3, and the rutile phase of GeO2.[12] We believe that our pressure and temperature revised in the cubic press are accurate, and the time of pressure and temperature increasing is so sufficient that the result is reliable. Comparing these conclusions, we can find that ZGO undergoes phase transformation from phase I to II at lower pressure and temperature. With increasing pressure and temperature, it is believed to bring about the decomposition reaction, but the ultimate crystal structure is diverse, which is relevant to the pressure and temperature. At ambient temperature, the phase transformation of ZGO is not observed up to 24 GPa and the phenomenon of PIA is found as the pressure is close to 10 GPa. However, ZGO can undergo the transformation from phase I to phase II above 1200 °C, to phase III around 1500 °C, respectively. Our result suggests that the mechanism of PIA is connected with kinetic hindrance.
3.2. The mechanism of PIA in ZGOThe PIA has been extensively studied in many materials.[15,17,19,21] Concurrently, the mechanism of PIA has been debated through the ages. A few factors such as framework structures, insufficient thermal energy, non-hydrostatic condition, increase in the coordination number and crystalline anisotropy have been considered to underlie PIA.[13,14,16,22] It is generally believed that the PIA easily arises from insufficient thermal energy in such a poly-tetrahedral packing structure.[16] PIA in the material of ZGO also contributed to the GeO4 and ZnO4 tetrahedron framework structure.
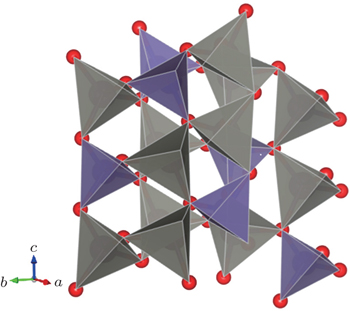
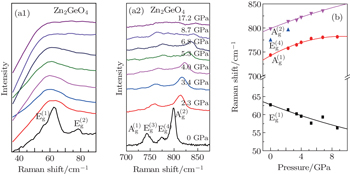
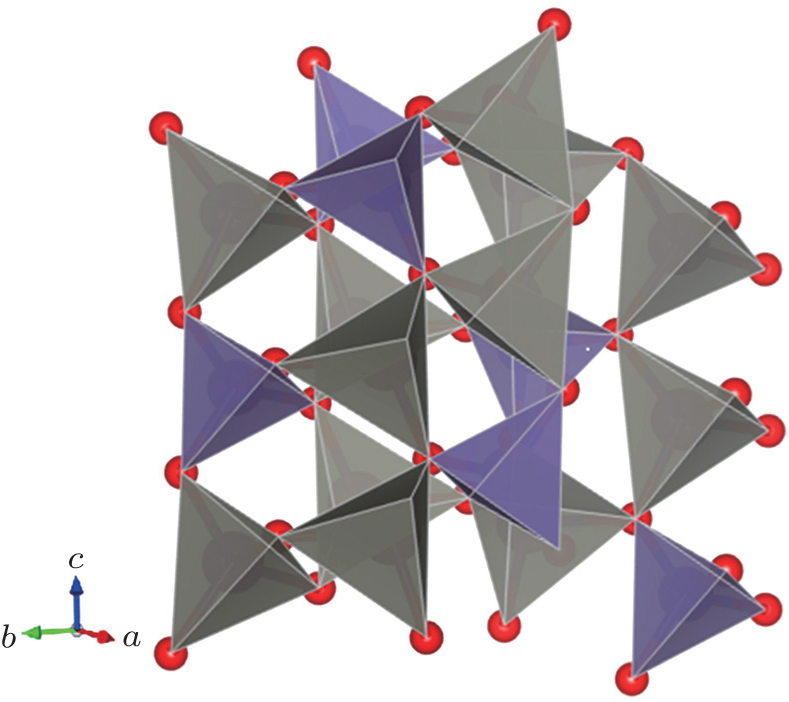
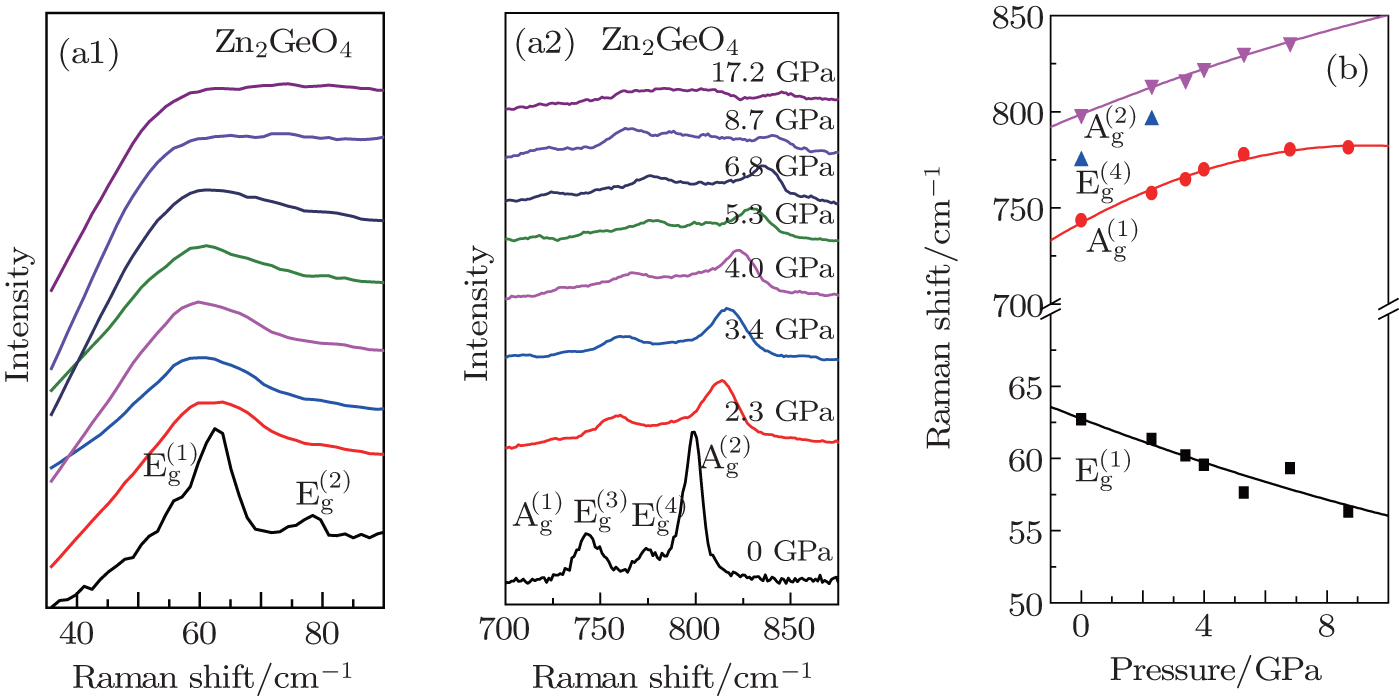
Figure 4 displays the crystal structure of hexagonal ZGO, which consists of GeO4 and ZnO4 tetrahedron. They are linked together by an oxygen atom, forming a three-dimensional tetrahedral network with large openings. They are alternate permutation, every oxygen atom links two asymmetric Zn atoms and one Ge atom. The high-pressure Raman spectra of ZGO display how to bring out the amorphous state in the poly-tetrahedral packing structure under high pressure (Figs. 5(a) and 5(b)). The highest pressure applied is 17.2 GPa. The hexagonal ZGO belongs to the point group-3, Ag, Eg, Au and Eu modes response the symmetry of the lattice vibration, Ag and Eg modes with the quadratic basis function are Raman active.[20] The Raman spectrum of ZGO displays a complex behavior with the vibrational modes of O–Zn–O, O–Ge–O, and Zn–O–Ge primarily.[18] The increase in band width and a frequency shift of phonon are discussed as the effect of the pressure. The analysis of the band width indicates that the ZnO4–GeO4 mode at 63 cm−1 and 78 cm−1, the Zn–O–Ge stretching mode at 743 cm−1, and the Zn–O–Ge bending mode at 776 cm−1 undergo order-disorder ahead of the O–Ge–O stretching mode at 801 cm−1 with increasing pressure.[20] Especially, the band width increasing and the phonon frequency shifting to the left of the ZnO4–GeO4 mode are different from the high-frequency vibration modes. This behavior is possibly attributed to the rupture of the linkage bond between the GeO4 and ZnO4 tetrahedron. The driving force for polyhedron tilting in the framework structure comes from the lower energy vibration modes.[16,23] Therefore, the preliminary deformation of ZGO is caused by the lower energy phonon softening.[14] In contrast to the low frequency region, it is obvious that the high frequency vibration modes at 743 cm−1, 776 cm−1, and 801 cm−1 shift to higher frequency, indicating that the vibration and energy gap of modes are enhanced. Upon further compression, the Raman peak of the O–Ge–O stretching mode at 801 cm−1 completely disappears at last, indicating that the inter-tetrahedral tilting Ge–O–Ge angle is varied, which results in the distorted tetrahedron. It is possible that the variation in bond angle instead of the decrease in the inter-atomic distances is the cause.[13]
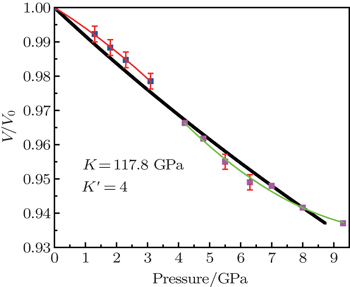
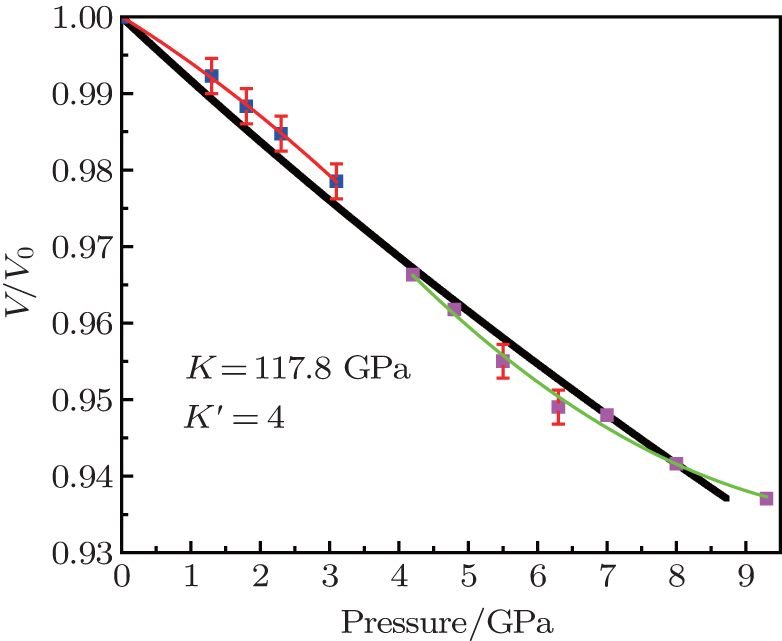
The speculation is also confirmed by analyzing the P–V/V0 (Fig. 6). The red and green fitting curves have similar slopes below 3.1 GPa and above 4.2 GPa, but the reduction ratio of V/V0 is discontinuous from 3.1 GPa to 4.2 GPa. We suspect that the crystal volume collapse is caused by the Zn–O–Ge bond rupture with increasing pressure, which is relatively serious. Because of the Zn–O–Ge bond linkages GeO4 and ZnO4 tetrahedron, its rupture can result in a more densely packed structure in the phase which also contains Ge and Zn in tetrahedron coordination. This process belongs to isomorphism phase transformation. Figure 2 shows that the diffraction patterns at 3.1 GPa and 4.2 GPa are uniform, demonstrating that the phase transformation comes from the same structure.
The pressure effect on the framework structure of ZGO brings about some disordered clusters due to Zn–O–Ge linkage bonds rupture, such as tetrahedron, hexahedron or octahedron unit cell. However, there is insufficient thermal energy to form a bond between two polyhedrons. In other words, the procedure of clusters nucleation and growth is hindered so that high-pressure equilibrium phase cannot be formed, maintaining the amorphous state. Here, the phase transformation can be performed at HP-HT. This is because these clusters are arranged in order by enough thermal energy under high pressure, so that the grown clusters probably long range orderly transform to cubic spinel and tetragonal spinel structure. In other words, high temperature can supplement the dynamic to rearrange these polyhedrons to form the phases. At low temperature and high pressures, the transformation to an amorphous phase is observed, which is a general phenomenon of the open-packed structure.[11,16]
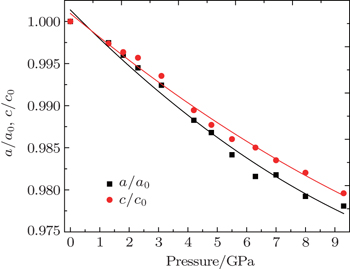
3.3. The compression properties of ZGO at high pressureThe compression behavior of ZGO under high pressure is reflected in the pressure–volume data plotted in Fig. 6. The present P–V data is analyzed with the following third-order Birch–Murnaghan equation of state (EoS):[24]
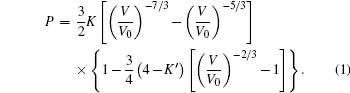
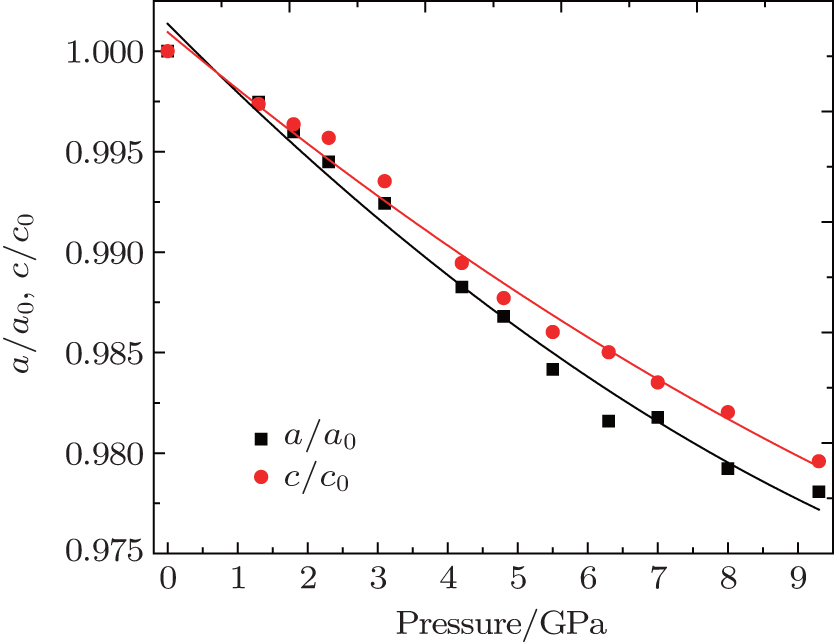
Here, V/V0 is the ratio of unit-cell volume compressed to 10 GPa comparing to ambient conditions. K is the bulk modulus, and K′ is the pressure derivative of K. The bulk modulus of material can be calculated by fixing the zero-pressure volume (V0) to its measured value. The EOS of the hexagonal ZGO is determined: V0 = 1671.4 Å, K = 117.8 GPa, K′ = 4, standard error of the bulk modulus is 11.7 GPa compared to standard EOS. The bulk modulus is an important parameter, which can present the material incompressibility. The pressure evolution of lattice parameters is shown in Fig. 7. A closer look at the compression behavior of the unit cell axes reveals that the crystals of ZGO are not obviously anisotropic, the compressibility of a-axis and c-axis is similar up to 3.1 GPa and the value decreases linearly. In order to understand the isotropic behavior below 3.1 GPa, it is essential to consider the crystal structure of the hexagonal phase. Since GeO4 and ZnO4 tetrahedrons are alternate permutations, the Zn–O–Ge linkages form a similar frame of a-axis and c-axis. As pressure increases, the compression behavior of the a-axis and c-axis is similar. Above 4.2 GPa, the compressibility becomes different. As a whole, the c-axis is more incompressible than the a-axis. The change is interpreted as a lattice collapse in the hexagonal ZGO. The direct result is that the framework structure becomes more distorted with increasing pressure.
4. Conclusion and perspectivesThe phase transition and compressibility of ZGO were investigated at pressure up to 24 GPa with room-temperature ADXRD and Raman. The HP-HT experiments were performed at cubic press to 5 GPa, 1500 °C. Pressure-induced irreversible amorphization of ZGO arises up to 10 GPa at room temperature, the hexagonal structure transforms to phase II–IV with increasing temperature, indicating that the PIA is connected with kinetic hindrance. Furthermore, the high-pressure Raman spectrum measurement illustrates that the instability of the framework structure mainly comes from tilting Zn–O–Ge and Ge–O–Ge bond angles. The compression curve of ZGO yields an averaged bulk modulus of 117.8±11.7 GPa. The material with these low frequency modes and highly flexible framework structure easily exhibits the PIA property. The study provides an effective method to make an amorphous substance using high pressure. Moreover, we can obtain different structure bulk solids of ZGO by changing the pressure and temperature at the cubic press.



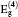
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 , Li Wen-Tao1, 2, Hu Qi-Wei1, Yan Xiao-Zhi1, 2, Lei Li1, Li Xiao-Dong3, He Duan-Wei1]
, Li Wen-Tao1, 2, Hu Qi-Wei1, Yan Xiao-Zhi1, 2, Lei Li1, Li Xiao-Dong3, He Duan-Wei1]