{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Hybrid density functional study on lattice vibration, thermodynamic properties, and chemical bonding of plutonium monocarbide
[Yang Rong1, 2, Tang Bin3, Gao Tao1, †,  , Ao BingYun4, ‡, ]
, Ao BingYun4, ‡, ]
, Ao BingYun4, ‡, ]
|
|


† Corresponding author. E-mail:
‡ Corresponding author. E-mail:
Project supported by the National Natural Science Foundation of China (Grant Nos. 21371160 and 21401173).
Hybrid density functional theory is employed to systematically investigate the structural, magnetic, vibrational, thermodynamic properties of plutonium monocarbide (PuC and PuC0.75). For comparison, the results obtained by DFT, DFT + U are also given. For PuC and PuC0.75, Fock-0.25 hybrid functional gives the best lattice constants and predicts the correct ground states of antiferromagnetic (AFM) structure. The calculated phonon spectra suggest that PuC and PuC0.75 are dynamically stable. Values of the Helmholtz free energy ΔF, internal energy ΔE, entropy S, and constant-volume specific heat Cv of PuC and PuC0.75 are given. The results are in good agreement with available experimental or theoretical data. As for the chemical bonding nature, the difference charge densities, the partial densities of states and the Bader charge analysis suggest that the Pu–C bonds of PuC and PuC0.75 have a mixture of covalent character and ionic character. The effect of carbon vacancy on the chemical bonding is also discussed in detail. We expect that our study can provide some useful reference for further experimental research on the phonon density of states, thermodynamic properties of the plutonium monocarbide.
Plutonium monocarbide has been a subject of intense interest in recent years, owing to its applications in advanced types of fast reactors. Compared with oxide fuels, it has many advantages. First, its very good thermal conductivity fits the important technological requirements for advanced reactors for higher operating temperatures. Second, plutonium monocarbide with the rock-salt structure is stable in a large domain of nonstoichiometry at high temperature, and its fusion temperature is high. Finally, high burn-up value (10% or more) can be obtained, since plutonium carbides contain a high concentration of metallic elements. These advantages can explain this interest in plutonium monocarbide. As is well known, plutonium monocarbide is a non-stoichiometric compound with the fcc Nacl-type structure.[1] PuC easily forms a defect structure with some of its carbon atom lattice sites vacated. We select PuC0.75 as a defect structure to be investigated. In order to properly utilize carbide fuels in a reactor, their phase relationships and physical properties must be well understood. Of all the physical properties, the need for reliable thermodynamic data on carbide fuels is well recognized by both the engineer and the scientist. The vibrational properties are critical for understanding phase stability and thermal transport. So this work is concerned with the vibrational, thermodynamic properties of plutonium monocarbide (PuC and PuC0.75). Additionally, the investigations of chemical bonding properties of PuC and PuC0.75 from theoretical viewpoint are indispensable to their applications in the nuclear industry.
Experimental studies on the plutonium-bearing materials are challenging because of their toxic and radioactive nature. Thus, theoretical predictions appear to be especially important for plutonium monocarbide. However, the 5f-electron localization phenomena pose a considerable challenge to the theoretical research. To correctly describe the strong electron–electron interactions of f-electron systems, the traditional density functional theory (DFT), DFT plus the Hubbard U[2,3] (DFT + U), dynamical mean field theory (DMFT),[4] and hybrid density functional[5,6] have been developed. DFT + U which has been widely used depends on the Hubbard correction term. The value of Ueff must be tuned to obtain a better description of different properties. In Pu-based compounds, a value of 4 eV for Ueff was generally adopted.[7–9] Hybrid functional method does not contain any system-dependent parameters, but it needs high computational resources and times. In this work, we present results by using hybrid functionals and DFT + U.
Comparing with plutonium oxides, relatively few studies focus on plutonium carbides. Among plutonium carbides, PuC is most studied, and the studies on non-stoichiometric compound are more scarce. Experimentally, the data of structural parameters can be obtained. Experimental work[10] revealed that plutonium monocarbide undergoes a magnetic phase transition at ≈ 100 K, leading to a simple antiferromagnetic structure with Pu along the (0.0,1) direction. The observed valence-band spectra of Pu carbides were obtained by Gouder et al.[11] The heat content (enthalpy) of PuC above 298 K was measured with an isothermal drop calorimeter by Kruger and Savage.[12] In theory, some progress has been made in the ground-state properties of plutonium monocarbide. The structural, electronic and magnetic properties of PuC were investigated by Wen et al.[9] Havela et al.[13] studied the magnetic properties of PuC by LDA + U calculations. Petit et al.[14] investigate the ground-state valency configuration of Pu ions in PuC by using the self-interaction corrected local spin-density approximation. However, to our knowledge, no data of phonon spectrum are available in the literature. The accurate determination of phonon spectrum can help us to acquire thermodynamic properties such as the Helmholtz free energy ΔF, internal energy ΔE, entropy S, and constant-volume specific heat Cv. In addition, the investigation of chemical bonding has not been included in the previous studies. Hence, we calculate the lattice vibrations, thermodynamic properties and chemical bonding of PuC and PuC0.75. Our investigations, especially for nonstoichiometric compound (PuC0.75) are of great significance.
In this work, we first investigate the crystal structures and magnetic properties of PuC and PuC0.75, and compare the results with experimental data to check whether hybrid functional method is suitable for PuC and PuC0.75. Then, phonon-dispersion curves of PuC and PuC0.75 are obtained, the Raman and infrared active modes at Γ point are further assigned and presented. Next, the thermodynamic properties including ΔF, ΔE, S, and Cv are predicted for PuC and PuC0.75. And finally, the chemical bonding properties of PuC and PuC0.75 are discussed in detail.
The rest of this paper is organized as follows. In Section 2 we describe our computational method and details, and the results are presented and discussed in Section 3. This paper is ended with conclusions in Section 4.
All our calculations are performed by using the full potential linearized augmented plane wave (FP-LAPW) method as implemented in the all electron WIEN2K code.[15,16] In the exchange–correlation functional used are GGA Perdew–Burke–Ernzerhof (PBE) formalism[17] and the local spin-density approximation (LSDA).[18] In the LSDA + U[19] calculations, the effective Hubbard U is 4.00 eV, which is used by Wen et al.[9] In the hybrid scheme, the PBE0 and Fock-0.25 hybrid functionals are employed. For PBE0 hybrid functional,[20,21] the exchange-correlation energy is
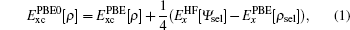
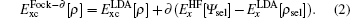
In our calculations of PuC, a supercell consisting of 4 Pu atoms and 4 C atoms is used. When one C atom is replaced by a vacancy, it forms C-deficient compound PuC0.75. We should mention that the experimentation[1] confirms that the C-deficient compound has the fcc Nacl-type structure.
The muffin-tin radii have been chosen to be 2.3 a.u. and 1.6 a.u. (The unit a.u. is short for atomic unit) for Pu and C atoms, respectively. The parameter RMTKMAX = 8.5, where RMT is the smallest atomic sphere radius and KMAX is the plane wave cutoff parameter. The Brillouin zone is sampled with 100 k-points. The charge convergence is set to be 0.001 e, the total energy convergence is set to be 0.001 Ry (1 Ry = 13.6056923(12) eV), and the force convergence is set to be 0.001 Ry/a.u. In the antiferromagnetic (AFM) calculations, we take the 1k (collinear) order. The spins of Pu atoms change sign along the oz axis.
We calculate the total energies for a set of different atomic volumes to determine the optimized lattice constants. The bulk modulus is obtained from fitting to Murnaghan’s equation of state.[23] The band gaps, the total magnetic moment and the differences in total energy between ferromagnetic (FM) and AFM states (ΔE = EFM − EAFM) are also obtained. All results are listed in Table
| Table 1. Complication of data on FM and AFM states of PuC and PuC0.75: lattice constant a0, total magnetic moment μtot, bulk modulus B0, ΔE = EFM − EAFM, and band gap (eV). The Hubbard parameter U is 4.0 eV. . |
The known experimental finding is that at room temperature the lattice parameter increases from a value of 4.953 Å to 4.973 Å as the carbon content increases from the hypo-to hyper-stoichiometric composition.[24–29] For stoichiometric PuC, the measured lattice parameter is 4.965 Å.[24] From Table
Of all the methods, Fock-0.25 yields the very accurate lattice constants. For example, for PuC in the AFM state, the value 4.961 Å deviates from the experimental data only by 0.004 Å. But PBE0 severely overestimates the lattice parameter in FM (or AFM) state. This indicates that GGA (PBE) is not suitable for plutonium monocarbide. When a C atom is removed from PuC to form C-deficient compound PuC0.75, a decrease of lattice constant is found in Table
As expected, the lattice parameter increases as long as the electron correlation (U or hybrid functional) is included. When Hubbard U increases from 0 to 4 eV, the localization of the 5f electrons of Pu will be enhanced, while the cohesion of the crystal is further reduced, which leads to the increase of the lattice constant. Moreover, we consider the effects of spin-orbit coupling (SO). When SO is included, the lattice parameter is reduced slightly in LSDA + U + SO/Fock-0.25 + SO framework compared with that in the LSDA + U/Fock-0.25, which is similar to the Pu–H systems calculated by Guo et al.[8] From the energy differences between the FM and AFM states in LSDA + U + SO/Fock-0.25 + SO framework, the ground states of PuC and PuC0.75 are also AFM. So spin–orbit coupling has little effect on crystal structure and magnetic properties of PuC and PuC0.75. Furthermore, the lattice constants of PuC and PuC0.75 in Fock-0.25 are much closer to the experimental values than those obtained by LSDA + U + SO/Fock-0.25 + SO. Thus, SO is not taken into account in the following calculations.
From our calculated band gaps, both PuC and PuC0.75 are conductors. It is true since plutonium monocarbide is metal. Finally, we compare our results with other theoretical values (HSE[9] and SIC-LSD[14]). We find that our calculated lattice parameters obtained by Fock-0.25 are much better than the others, suggesting that the advantage of the present method (Fock-0.25) over the others for plutonium monocarbide. For the bulk modulus B0, our calculated value 177.40 GPa from Fock-0.25 can be compared with the result 172.00 GPa from SIC-LSD.[14]
On the whole, Fock-0.25 gives the best structural properties and predicts AFM phase, which supplies the safeguard for the following calculations of vibrational and thermodynamic properties of PuC and PuC0.75. For comparison, LSDA + U calculations will be also performed in the following studies. While PBE0 is not adopted in the following calculations, since the optimized lattice parameters are not reproduced well by PBE0.
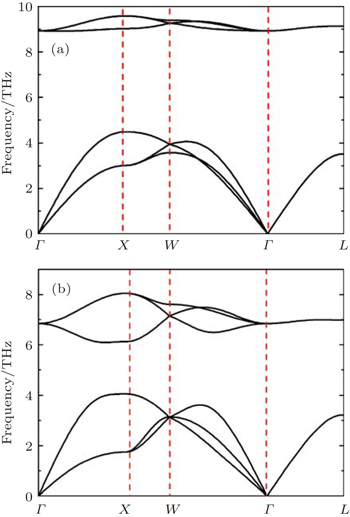
Our phonon calculations are performed by the small displacement method.[30] Since the space groups of PuC and PuC0.75 are Fm-3m (225), PuC and PuC0.75 belong to Oh point group. The high-symmetry points selected in the calculations are Γ(0, 0, 0), X (0.5, 0, 0.5), W (0.5, 0.25, 0.75), and L (0.5, 0.5, 0.5). For stoichiometric PuC, there are two atoms in a primitive cell. Thus, this system contains three acoustic modes and three optical modes, that is, two optical transverse branches and one optical longitudinal branch, two acoustic transverse branches and one acoustic longitudinal branch are included. The phonon dispersion curves of PuC obtained from Fock-0.25 and LSDA + U are shown in Figs.
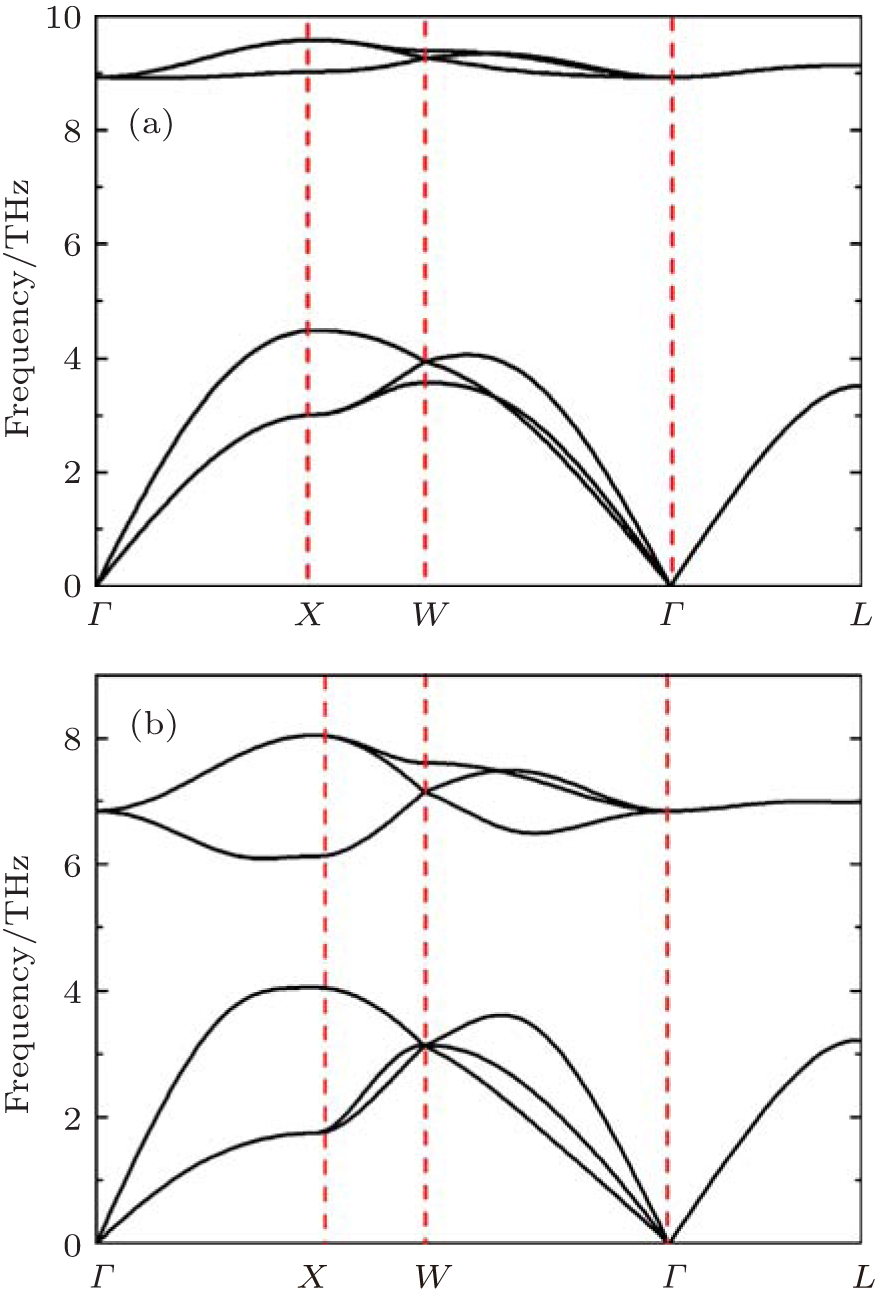 | Fig. 1. Calculated phonon dispersion curves from (a) Fock-0.25 and (b) LSDA + U for PuC. |
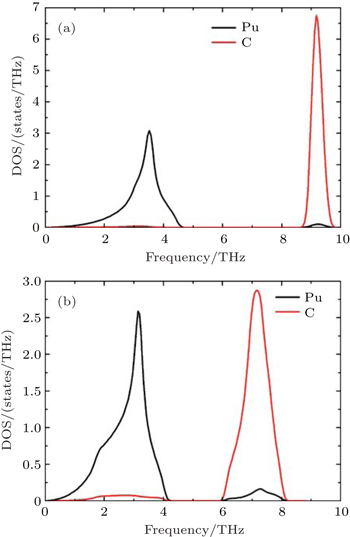
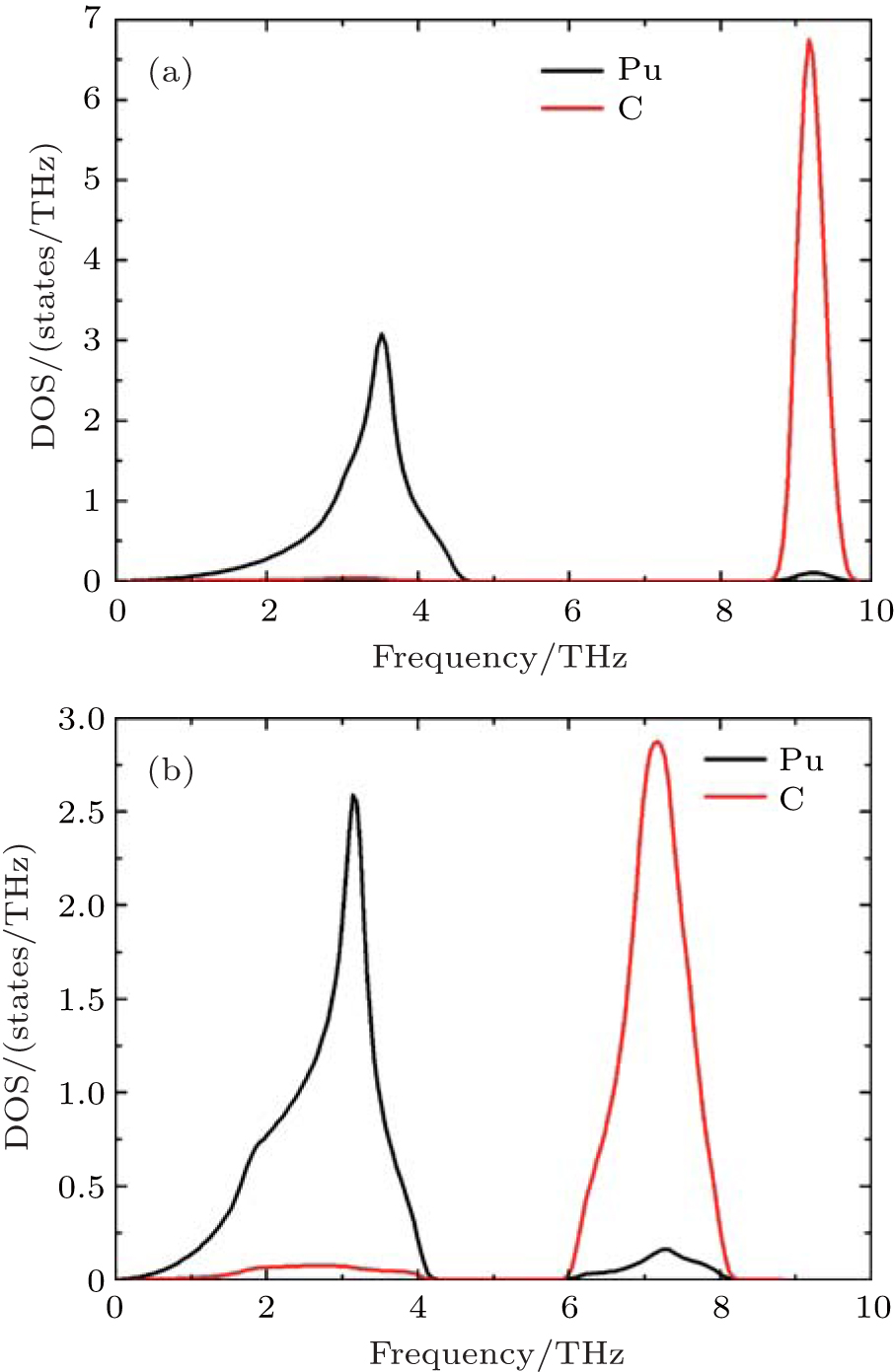 | Fig. 2. Calculated phonon partial densities of states (PPDOSs) from (a) Fock-0.25 and (b) LSDA + U for PuC. |
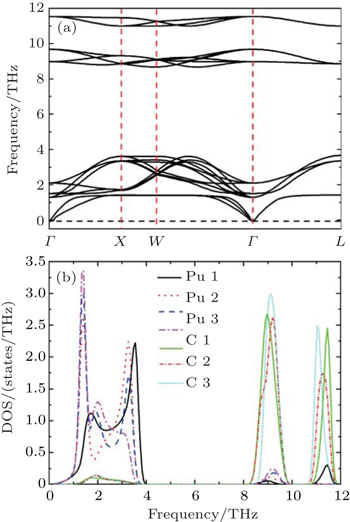
Now we discuss the lattice dynamical properties of PuC0.75. For simplicity, we only show the results from Fock-0.25. From Fig.
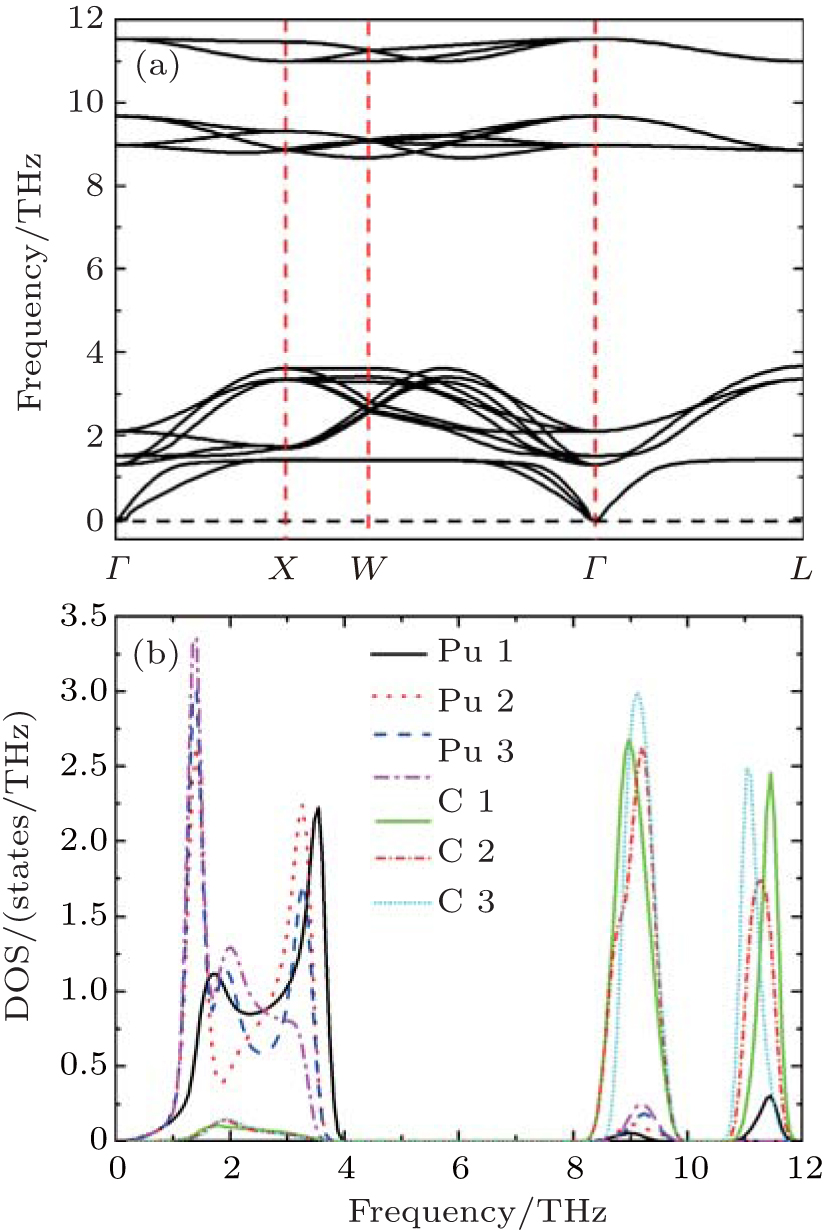 | Fig. 3. Calculated (a) phonon dispersion curves and (b) partial density of phonon states (PPDOSs) from Fock-0.25 for PuC0.75. |
| Table 2. Calculated phonon frequencies (in cm−1) and the assignment of the infrared-active and silent modes at Γ point of PuC0.75. . |
To our knowledge, there are currently no experimental or theoretical data for the vibrational properties of PuC and PuC0.75. We hope our results can provide useful reference for the further research on the vibrational properties of PuC and PuC0.75.
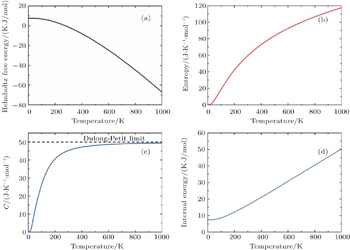
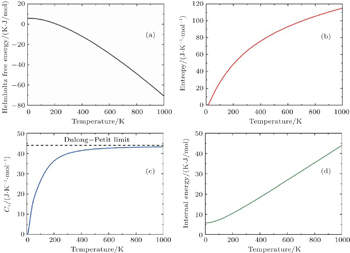
Thermodynamic properties of solid materials can be determined by the calculated phonon density of states in the quasiharmonic approximation (QHA). The expressions of the Helmholtz free energy ΔF, internal energy ΔE, entropy S, and constant-volume specific heat Cv are given, respectively, by[31]
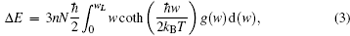
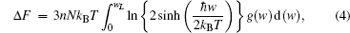
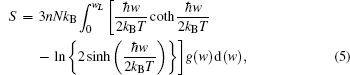
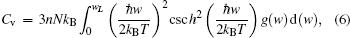
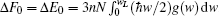
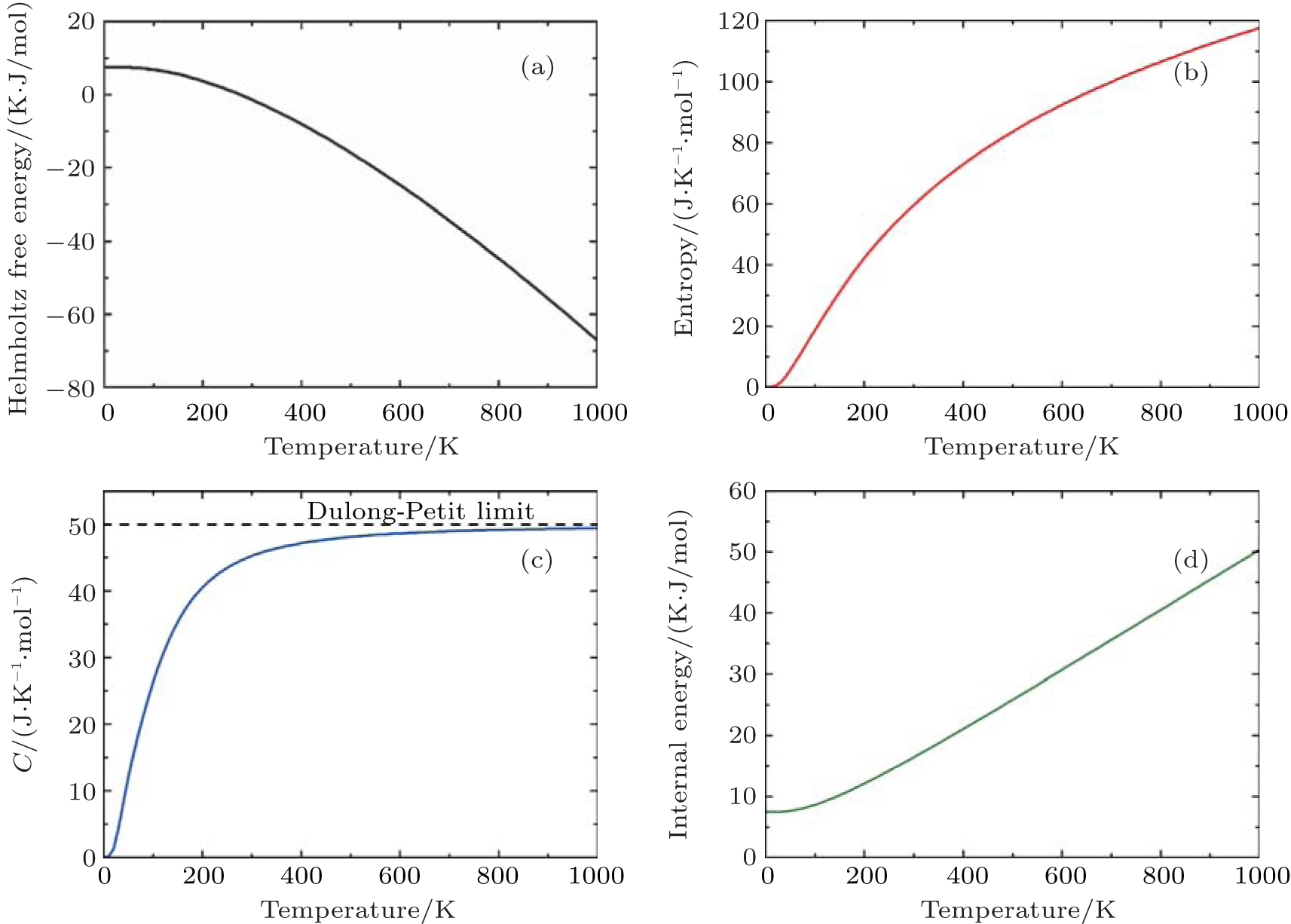 | Fig. 4. Calculated (a) the Helmholtz free energy ΔF, (b) the entropy S, (c) the constant-volume specific heat Cv, and (d) the internal energy ΔE of PuC in the Fock-0.25 framework. |
The observed changes in enthalpy of PuC with temperature rising are listed in Table
| Table 3. Calculated values of H–H298 (K·J/mol) and the corresponding experimental data of PuC. . |
Listed in Table
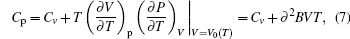
| Table 4. Our calculated values of ST–S298 (J/(K/mol)) and heat capacity (J/(K/mol)) of PuC, along with other theoretical values. . |
For PuC0.75, we also investigate its thermodynamic properties. In Table
| Table 5. Our calculated values of H–H298 (K·J/mol), S (J/(K/mol)), and Cv (J/(K/mol)) of PuC0.75. . |
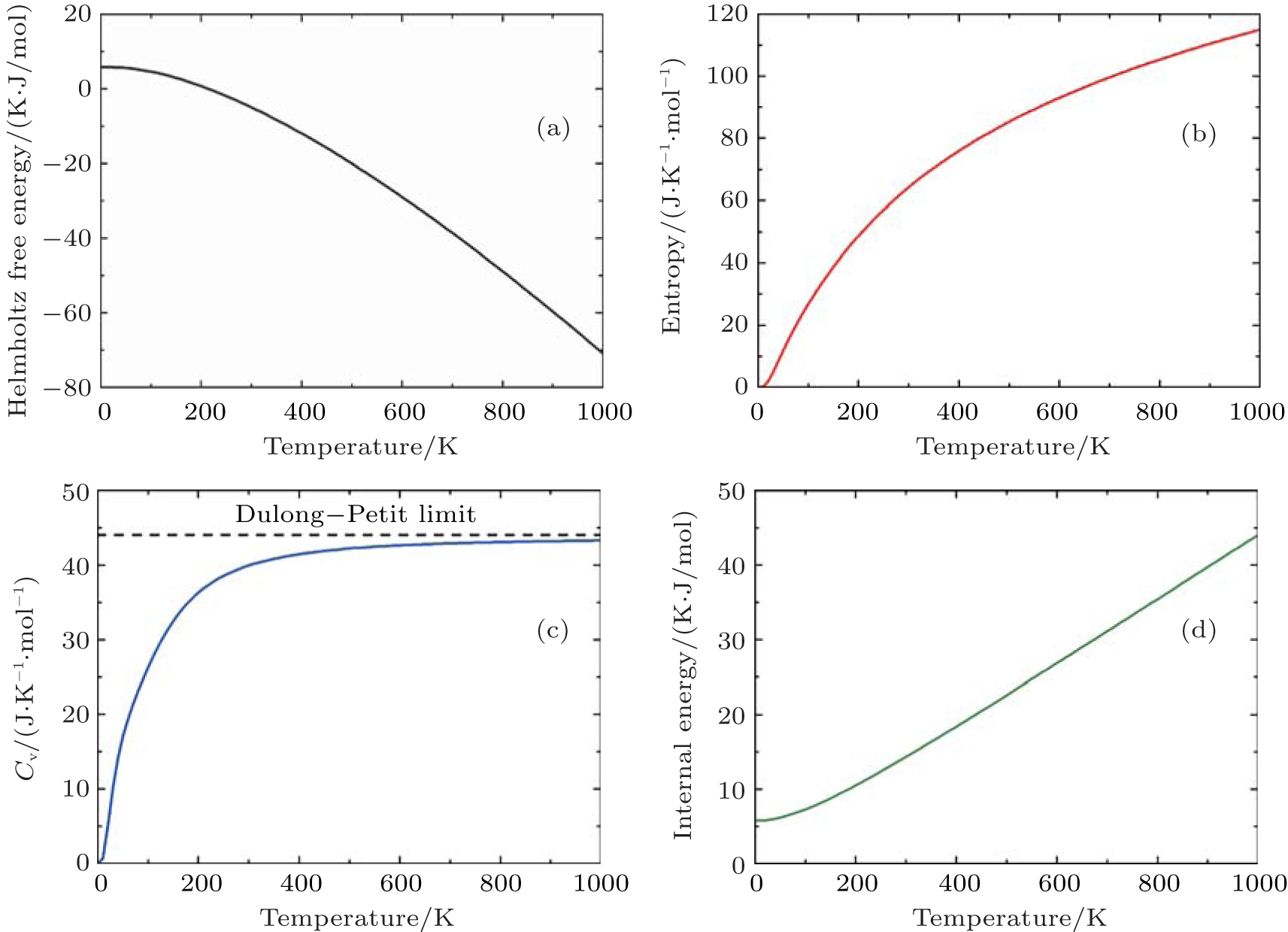 | Fig. 5. Calculated (a) the Helmholtz free energy ΔF, (b) the entropy S, (c) the constant-volume specific heat Cv, and (d) the internal energy ΔE of PuC0.75 in the Fock-0.25 framework. |
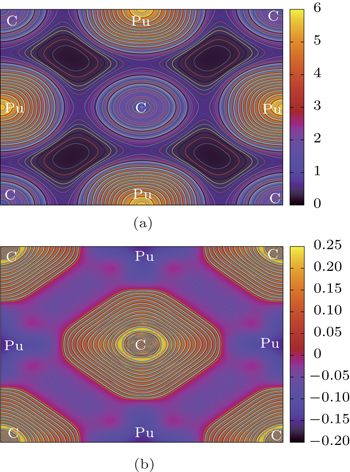
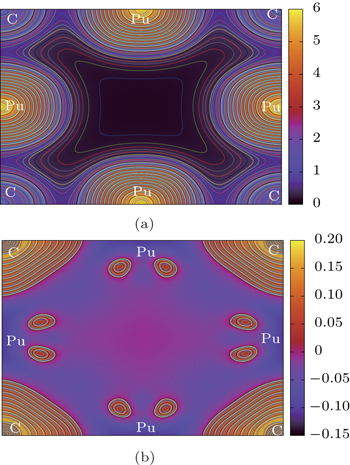
In this section, we will analyze the Pu–C bonds of PuC and PuC0.75. The charge densities and the difference charge densities along the (001) plane of PuC and PuC0.75 are shown in Figs.
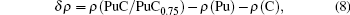
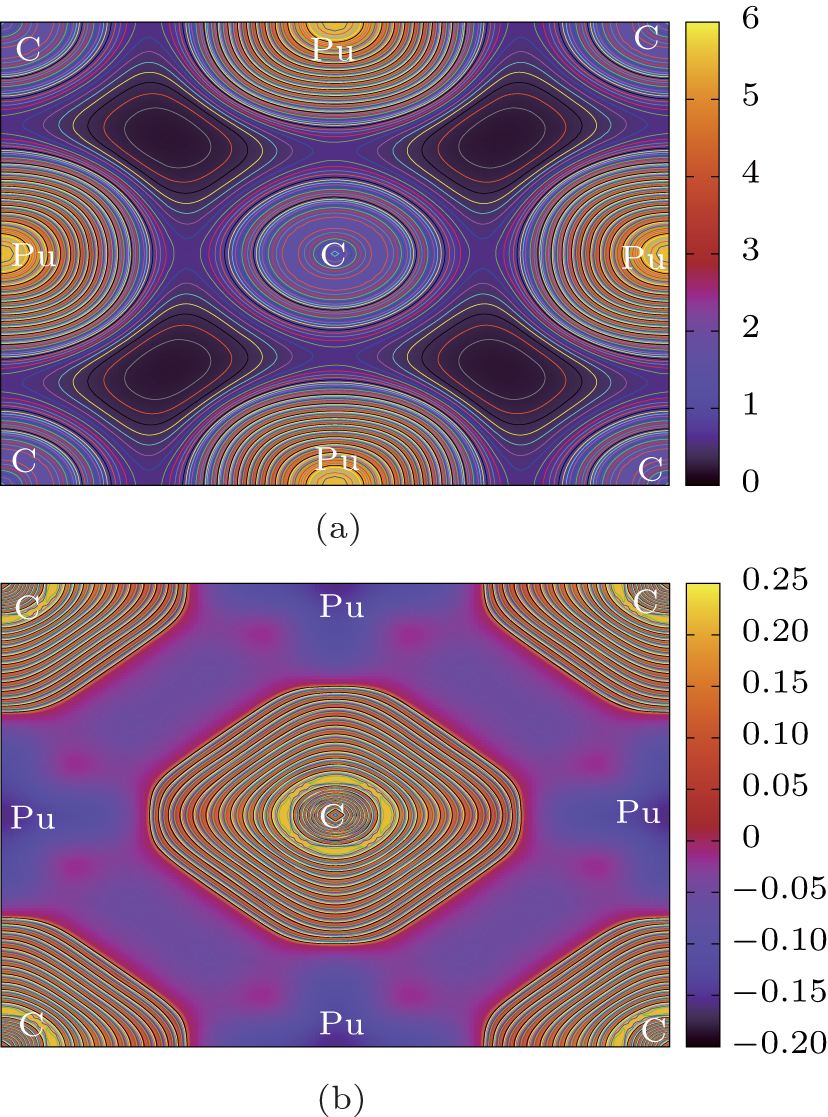 | Fig. 6. Contour plots of (a) charge density and (b) charge density difference along the (001) plane in Fock-0.25 calculations for PuC. |
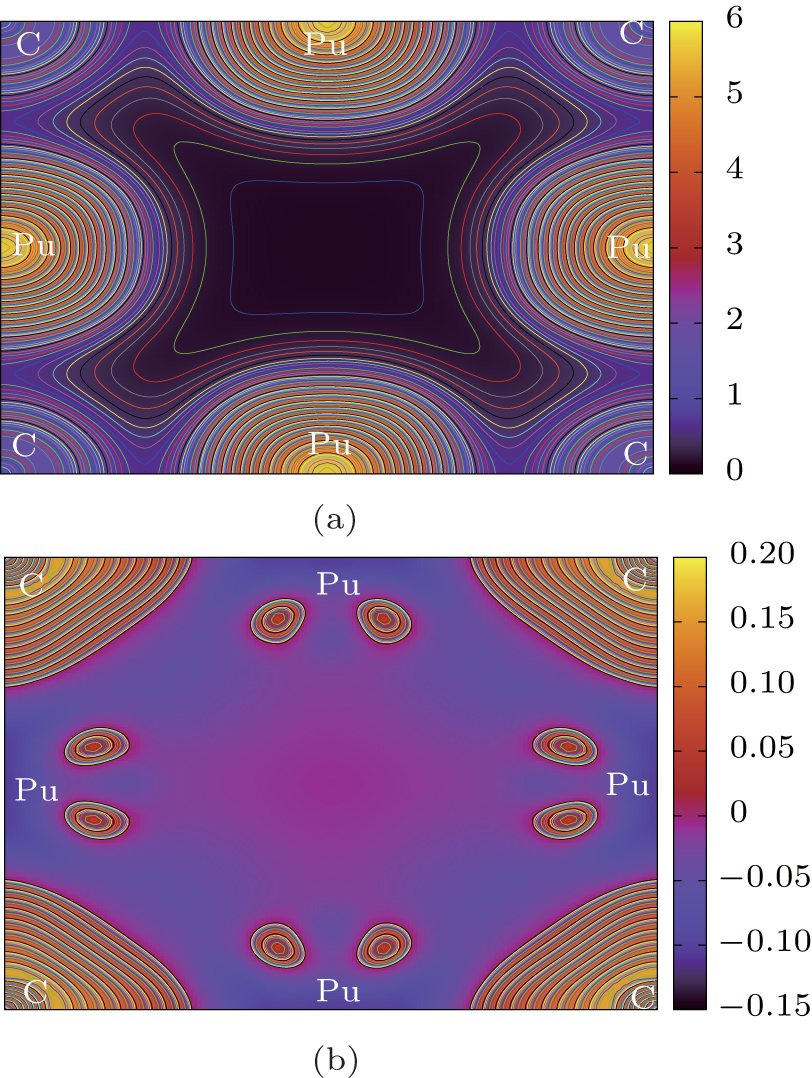 | Fig. 7. Contour plots of (a) charge density and (b) charge density difference along the (001) plane in Fock-0.25 calculations for PuC0.75. |
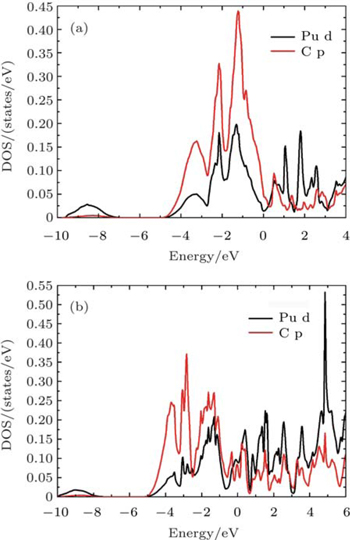
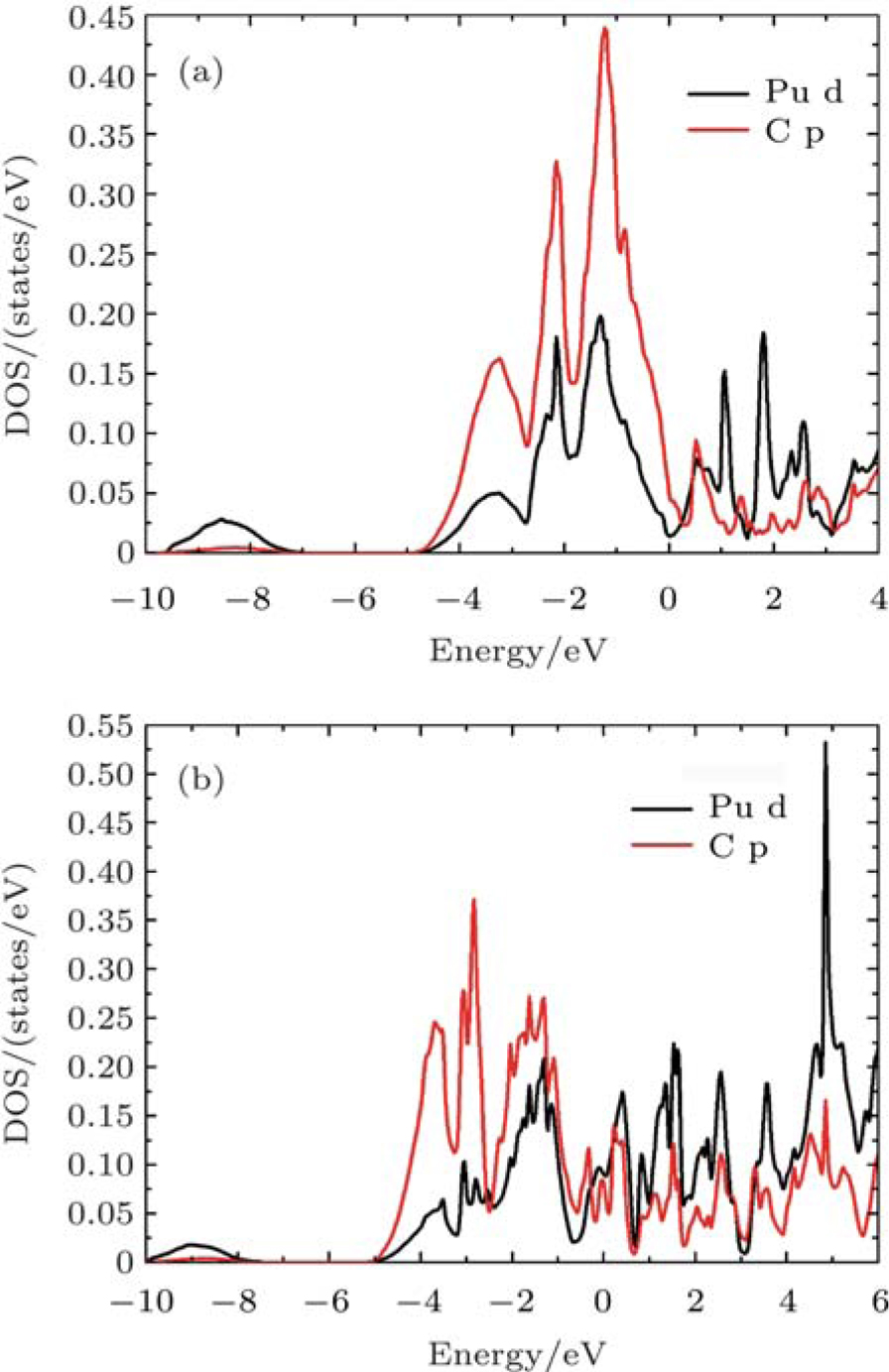 | Fig. 8. Partial densities of states for the Pu d and C p of (a) PuC (b) PuC0.75 in Fock-0.25 calculations. |
The charge transfer between the Pu atom and the C atom is quantified by the Bader charge analysis.[42] As is well known, Pu atom has 16 electrons outside the core, and C atom has 4 electrons outside the core. All results are listed in Table
| Table 6. Calculated bader charges (QB) and bader volumes (VB) for AFM PuC and AFM PuC0.75 in Fock-0.25 framework. . |
According to the charge densities and the difference charge densities, the partial densities of states, the Bader charge analyses for PuC and PuC0.75, we can reach the following conclusion. (i) Mixtures of covalent and ionic character are present in the Pu–C bonds of PuC and PuC0.75. (ii) The ionic character is stronger than the covalent character in the PuC system. (iii) Near the vacancy, Pu ions in PuC0.75 are ionized somewhat less than Pu ions in PuC, and the ionic character is weaker than the covalent character in the PuC0.75 system. (iv) Pu–Pu bonds near the vacancy are strengthened.
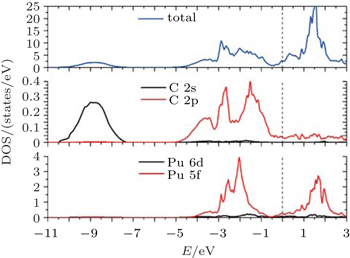
The observed valence-band spectra[11] of PuC1−x are characterized by the fingerprint of a triplet of sharp features, located at (or slightly below) the Fermi energy, 0.50 eV and 0.85 eV. And the spectra are dominated by the 5f character. So at the end of this section, we give the total and partial densities of states for PuC0.75 in Fock-0.25 scheme for comparison. From Fig.
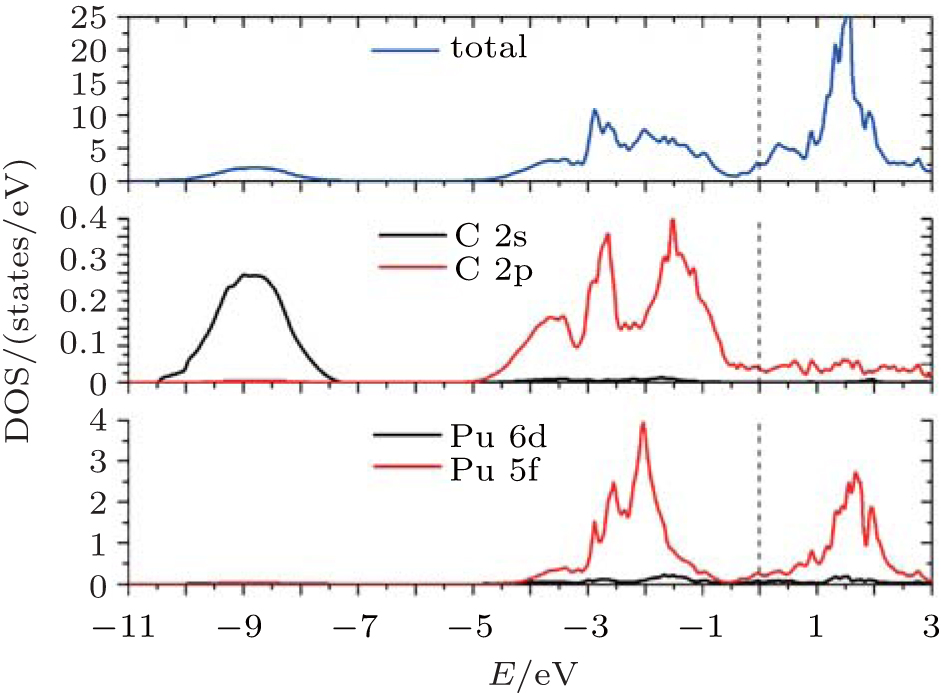 | Fig. 9. Total and partial densities of states for PuC0.75 in Fock-0.25 calculations. |
The lattice vibrations, thermodynamic properties and chemical bondings of PuC and PuC0.75 are systematically studied by using first-principle calculations. Since PuC and PuC0.75 are strongly correlated materials, DFT + U and hybrid density functional which replaces part of the DFT exchange by 25% exact HF exchanges are employed. The optimized lattice constants are in good agreement with known experimental data. LSDA + U and Fock-0.25 successfully predict the correct ground states of AFM PuC and PuC0.75 reported in experiments. We adopt the LSDA + U and Fock-0.25 to study the phonon dispersion curves and the corresponding PPDOSs of PuC and PuC0.75. The infrared active and silent modes at Γ point are further assigned. On the basis of the phonon DOS, we also systematically predict the thermodynamic properties of PuC and PuC0.75. The calculated values of H–H298, ST–S298, and S are in reasonably agreement with the experimental or theoretical values. The present results indicate that our computational methods are feasible and our prediction of the dynamical properties is believable. Through the charge densities, the difference charge densities, the partial densities of states, and the Bader charge analyses, we also carry out systematical studies of chemical bonding. We find that the mixtures of covalent and ionic character are present in the Pu–C bonds of PuC and PuC0.75. That one C atom is replaced by a vacancy leads to the growing of plutonium states in this region and their delocalization toward Pu-Pu bonds near the lattice defect. We expect that our work can provide some valuable information about the plutonium monocarbide for further experimental investigations.
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 | |
| 22 | |
| 23 | |
| 24 | |
| 25 | |
| 26 | |
| 27 | |
| 28 | |
| 29 | |
| 30 | |
| 31 | |
| 32 | |
| 33 | |
| 34 | |
| 35 | |
| 36 | |
| 37 | |
| 38 | |
| 39 | |
| 40 | |
| 41 | |
| 42 |