1. IntroductionSince the discovery of graphene, two-dimensional (2D) materials have attracted a great deal of attention by the research community due to the superiority of their excellent physical and chemical properties utilized in next generation nano-electronic and nano-optoelectronic devices.[1–4] Nevertheless, the incompatibility with the current silicon-based electronic technology hinders the potential application of the graphene. Silicene, a silicon analogue of graphene, has also attracted increasing attention for its novel chemical and physical properties,[5] such as the electric response,[6,7] ferromagnetism,[8,9] half-metallicity,[10,11] quantum Hall effect,[12] giant magnetoresistance,[13] and superconductivity.[14] Unlike planar graphene, the stablest silicene prefers a low-buckled (LB) structure, which is attributed to the mixing of its sp2/sp3 hybridized orbitals. The electronic structure of silicene is quite similar to that of graphene, exhibiting a semi-metallic character, in which the electrons at the Fermi level behave like massless Dirac fermions, thus leading to an extremely high carrier velocity.[15] As a consequence, the nano-devices fabricated by silicene will be more conveniently incorporated into the current silicon-based microelectronic industry without using carbon instead of silicon.
Adsorbing foreign atoms on silicene has significant potential applications. The adsorption of metal atoms provides a route to modifying the electronic properties of silicene. Ni et al. theoretically predicted that for silicene a band gap can be opened at the Dirac point with n-type doping by Cu, Ag, and Au adsorption, p-type doping by Ir adsorption, and neutral doping by Pt adsorption.[16] Peeters et al. studied the adsorption characteristics of alkali, alkaline-earth, and transition metal adatoms on silicene.[17] The adsorptions of Li, Na, and K result in the metalization of silicene, while the adsorptions of Be, Mg, and Ca turn silicene into a narrow gap semiconductor. Depending on the adatom type and atomic radius, the silicenes with adsorptions of transition metals can exhibit metal, half-metal, and semiconducting behavior respectively. Also, silicenes with adsorbed metal atoms, such as Li and K, are predicted to be promising host materials for hydrogen storage.[18,19] In addition, large adsorption energy and low diffusion energy barrier of Li atom on silicene suggest that silicene is also a suitable material for future anodes of lithium ion batteries (LIBs).[20] Note that the applications mentioned above need to avoid clustering the metal atoms when depositing metal atoms on silicene with high coverage. Therefore, the adsorption energy and diffusion energy barrier, which would affect the distribution of metal atoms on silicene, are important.
Up to now, silicene or silicene nanoribbons have been successfully fabricated on Ag,[21–27] Ir,[28] and ZrB2[29] substrates. Our previous study[30] showed that the O2 molecule dissociates on the silicene surface easily without overcoming any energy barrier, and the migration and desorption of O atoms are relatively difficult at room temperature due to the strong Si–O bonds. Therefore, the freestanding and pristine silicene is presumed to be unstably existent in the ambient condition. In contrast to the pristine silicene, full-hydrogenated silicene, so called silicane (SiH), may be realized more easily. Actually, silicene was initially synthesized by chemical exfoliation of calcium silicide (CaSi2),[31] where the silicon nanosheet is a monoatomic layer of silicon with a hexagonal crystal structure, and the surface of the monoatomic layer is capped with hydrogen. That is to say, the prepared silicene by using chemical exfoliation is in fact the silicane. Therefore, the study of the hydrogenated silicene should be essentially meaningful. Hussain et al.[32] and Yang et al.[33] have studied hydrogenated silicene functionalized with alkali and alkaline-earth metals for efficient hydrogen storage. In their studies, however, the metal atoms bind with silicane through substituting the hydrogen atoms with metal atoms. Strictly speaking, this is the interaction between metal atoms and defective silicane, not the adsorption of metal atoms on the pure silicane.
In order to explore the adsorption and diffusion behaviors of alkali and alkaline-earth metal atoms on the pure silicane, in this work, we calculate the adsorption energies and diffusion energy barriers of the alkali (Li, Na, K), alkaline-earth (Be, Mg, Ca) metal atoms on the silicane surface by using first-principles calculations. We also compare the results of the metal atoms on silicane with those on silicene. Our results show that the adsorption energies of all metal atoms considered in our calculations on the silicane are significantly smaller than those on silicene, and also smaller than the cohesive energy of metal. Furthermore, the diffusion energy barriers of the alkali metal atoms on silicane are comparable to those on silicene, whereas the barriers of the alkaline-earth are essentially smaller than those on silicene. Combining the results of the adsorption energies and the results of the diffusion energy barriers, it would be found that the clustering would occur when depositing metal atoms on hydrogenated silicene with relative high coverage.
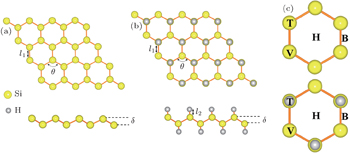
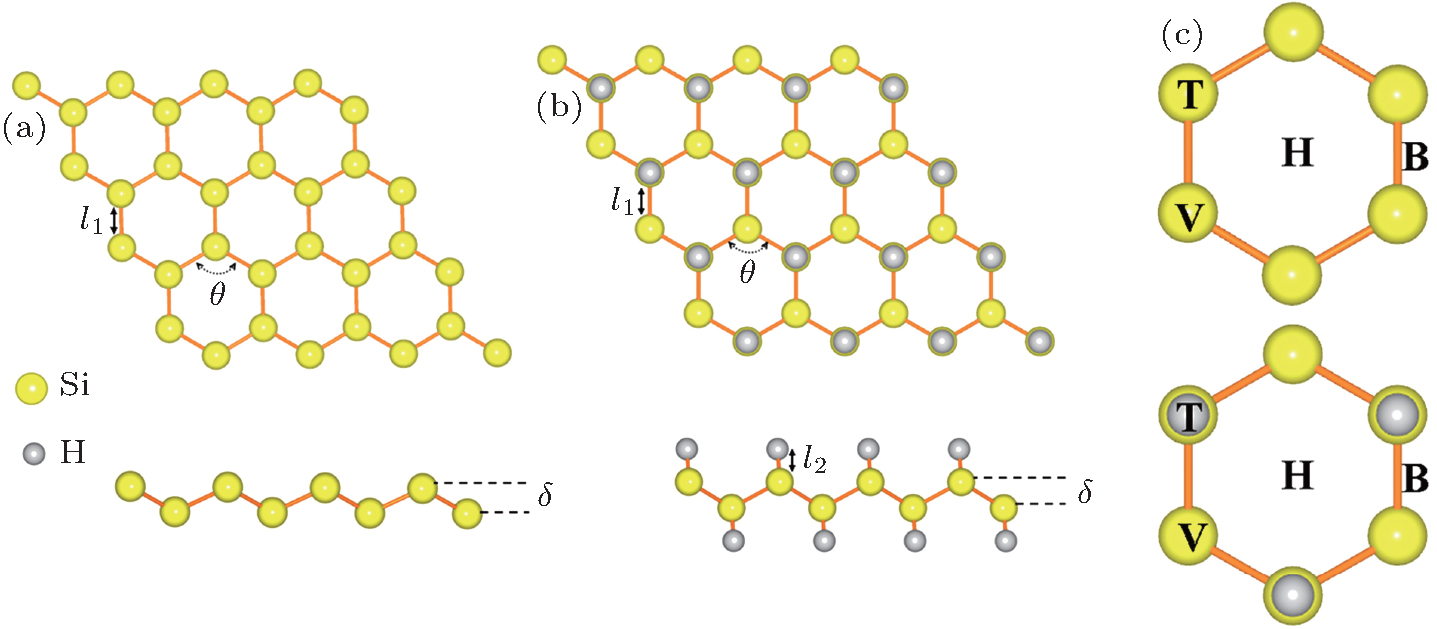
3. Results and discussionFirst, we examine the perfect structures of silicene and silicane. The optimized geometric structures are plotted in Fig. 1(a), and the corresponding structural parameters are listed in Table 1. The lattice constant (a), Si–Si bond length (l1) and buckling distance (δ) of silicene are 3.87, 2.28, and 0.45 Å, respectively, which are in good agreement with previously reported results.[7,40] For the silicane, our investigations mainly focus on the chair-like structure, which is proved to be the stablest configuration by Houssa et al.[41] Compared with the lattice parameters of silicene, the lattice constant and Si–Si bond length are slightly large, which are 3.89 Å and 2.36 Å, respectively. Especially, the buckling distance of silicane is 0.72 Å, significantly larger than that of silicene, which is most likely because the hydrogen adsorption induces the formation of sp3 orbital hybridization for Si atom. Our results for the silicane are consistent with previously reported results.[42] In addition, the optimized Si–H bond length is 1.5 Å.
Table 1.
Table 1.
 Table 1. Structural parameters of silicene and silicane. a represents the lattice constant of an optimized unit cell, l1 and l2 are the Si–Si and Si–H bond lengths, respectively. δ and θ are the buckling distance and bond angle respectively, which are marked in Fig. 1. .
|
a/Å |
l1/Å |
l2/Å |
δ/Å |
θ/rad |
| Silicene |
3.87 |
2.28 |
– |
0.45 |
116.20 |
| Silicane |
3.89 |
2.36 |
1.50 |
0.72 |
111.15 |
| Table 1. Structural parameters of silicene and silicane. a represents the lattice constant of an optimized unit cell, l1 and l2 are the Si–Si and Si–H bond lengths, respectively. δ and θ are the buckling distance and bond angle respectively, which are marked in Fig. 1. . |
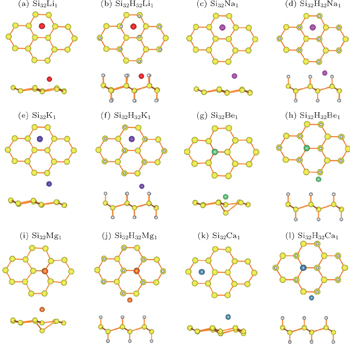
Next, the adsorption behaviors of alkali metal atoms (Li, Na, K) and alkaline-earth metal atoms (Be, Mg, Ca) are investigated. Considering the symmetries of the silicene and silicane substrates, four different adsorption sites, namely, above the center of hexagonal silicon rings (hollow site H), at the top of the upper silicon atoms or hydrogen atoms (top site T), at the top of the lower silicon atoms (valley site V), and at the top of the Si–Si bond (bridge site B), are taken into account for the atom adsorption. The corresponding adsorption sites are marked in Fig. 1(c). According to the total energy calculations, the stablest adsorption geometries are shown in Fig. 2. The corresponding structural parameters, adsorption sites and adsorption energies of the metal atoms are listed in Table 2.
In addition, the detailed total energies of silicene and silicane with adatoms at different initial adsorption sites are also provided in Table 3. Specifically, H sites are the ultimate adsorption sites for Li, Na, K, and Ca atoms on silicene, whereas V sites for Be and Mg atoms. Carefully examining the optimized structures of the silicene with the adsorptions of metal atoms, it is found that the silicene substrate basically keeps the original configurations for Li, Na, K, and Ca adsorption. Therefore, the average buckling distance (δ) values are all about 0.47 Å, which is slightly larger than that of perfect silicene. For the cases of Be and Mg, however, the Si atoms under the metal atoms are significantly pushed down after the adsorptions of Be and Mg metal atoms on silicene, which results in the relatively large distortion of the silicene. Interestingly, the distortion leads to the decreases of the average buckling distances down to 0.425 Å and 0.430 Å for Be- and Mg-adsorbed silicene, respectively. On the other hand, we investigate the adatom height (h), which is defined as the vertical distance between the adsorbed atom and the upper silicon atom. The adatom heights are 1.309, 1.922, and 2.515 Å for the alkali metal atoms Li, Na, and K, and 0.879, 1.645, and 1.696 Å for Be, Mg, and Ca alkaline-earth metal atoms, respectively.
Table 2.
Table 2.
Table 2. Calculated results for alkali and alkaline-earth metal atoms adsorbed on silicene and silicane. Hollow (H), bridge (B), valley (V) and top (T) represent the adsorption sites. h is the adatom height, and δ is the buckling distance. Ead is the adsorption energy of adatom. .
|
Site |
h/Å |
δ/Å |
Ead/eV |
| Si32Li1 |
H |
1.309 |
0.473 |
2.429 |
| Si32Na1 |
H |
1.922 |
0.475 |
1.848 |
| Si32K1 |
H |
2.515 |
0.474 |
2.109 |
| Si32Be1 |
V |
0.879 |
0.425 |
2.403 |
| Si32Mg1 |
V |
1.645 |
0.430 |
0.886 |
| Si32Ca1 |
H |
1.696 |
0.473 |
2.234 |
| Si32H32Li1 |
H |
1.397 |
0.741 |
1.084 |
| Si32H32Na1 |
H |
2.044 |
0.724 |
0.475 |
| Si32H32K1 |
H |
2.619 |
0.712 |
0.696 |
| Si32H32Be1 |
V |
3.711 |
0.716 |
0.049 |
| Si32H32Mg1 |
V |
4.114 |
0.726 |
0.057 |
| Si32H32Ca1 |
V |
4.646 |
0.717 |
0.088 |
| Table 2. Calculated results for alkali and alkaline-earth metal atoms adsorbed on silicene and silicane. Hollow (H), bridge (B), valley (V) and top (T) represent the adsorption sites. h is the adatom height, and δ is the buckling distance. Ead is the adsorption energy of adatom. . |
Next we discuss the adsorption energies of metal atoms on silicene. Our calculated adsorption energies of Li, Na, and K metal atoms, which are listed in Table 2, agree well with the results reported by Sahin and Peeters,[17] in which the adsorption energies of Li, Na, and K on silicene are 2.40, 1.85, and 2.11 eV, respectively. Compared with the corresponding values of Ni, etc.,[43] however, our results are somewhat large. Despite this, the differences in adsorption energy between Li and Na, or Na and K in our calculations are basically equal to those from Ni, etc. In addition, for the adsorptions of Be, Mg, and Ca atoms on silicene, the adsorption energies in our work are about 0.4 eV lower than those of previous results reported by Sahin and Peeters.[17] However, it is worth noting that both the difference in adsorption energy between Be and Mg and that between Mg and Ca in our calculations are almost the same as those in the results of Sahin and Peeters. Therefore, our results of the adsorption energies are reliable.
After the silicene is hydrogenated, thus forming silicane, the adsorption behaviors of metal atoms change a little. The adsorption sites of metal atoms are basically kept unchanged except for the case of Ca, in which the H site is changed into a V site. The buckling distance of silicane has little change after the atom adsorption. The adatom heights of Be, Mg, and Ca atoms on silicane are 3.711, 4.114, and 4.646 Å, respectively, each of which is significantly larger than that on silicene. However, the heights are close to those on silicene for the cases of Li, Na, and K. More importantly, the adsorption energies of all metal atoms on silicane are essentially lower than those on silicene. For a more convenient comparison, we arrange the adsorption energies of all metal atoms on silicane and silicene in one figure as seen Fig. 4(a). From Fig. 4(a), it is found that the basic trend of the adsorption energies of Li, Na, and K on siliance are similar to those on silicene except the different values of adsorption energies. For the adsorptions of Be, Mg, and Ca atoms on silicane, however, the adsorption energies are all below 0.1 eV, indicating the weak interaction between alkaline-earth metal atoms and silicane substrate.
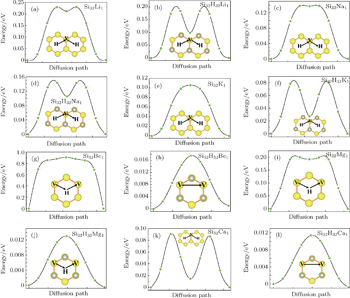
The diffusion behavior is another important issue for the adsorbed metal atoms. Therefore, we investigate the diffusion barriers of the metal atoms on different substrates, i.e., silicene and silicane, using the NEB method. The calculated energy profiles are plotted in Fig. 3, and the corresponding optimized diffusion paths are also shown as the inserts. When the metal atoms are adsorbed on silicene, the diffusion paths are from the H site to the nearest neighboring H site with passing the V site for Li, Na, K, and Ca atoms as shown in Figs. 3(a), 3(c), 3(e), and 3(k), whereas from the V site to the nearest neighboring V site with passing the H site for Be and Mg atoms, as seen in Figs. 3(g) and 3(i). For the cases of Li, Na, K, and Mg on silicane, the diffusion paths are similar to those on silicene as shown in Figs. 3(b), 3(d), 3(f), and 3(j). However, the diffusion paths for Be and Ca atoms are from the V site directly to the nearest neighboring V site, without passing the H site as shown in Figs. 3(h) and 3(k). Although the energy profiles for the diffusions of the metal atoms are plotted in Fig. 3, the diffusion barriers are also shown in Fig. 4(b) for the better comparison.
Table 3.
Table 3.
Table 3. Optimized total energies of silicene and silicane with adatoms at different initial adsorption sites. .
| Atom M |
Site |
Si32M1/eV |
Si32H32M1/eV |
| Li |
B |
−155.497 |
−281.910 |
|
H |
−155.704 |
−281.982 |
|
T |
−155.318 |
−281.130 |
|
V |
−155.498 |
−281.910 |
| Na |
B |
−155.006 |
−281.307 |
|
H |
−155.133 |
−281.383 |
|
T |
−154.869 |
−281.060 |
|
V |
−155.007 |
−281.306 |
| K |
B |
−155.315 |
−281.602 |
|
H |
−155.406 |
−281.617 |
|
T |
−155.222 |
−281.308 |
|
V |
−155.316 |
−281.598 |
| Be |
B |
−155.712 |
−280.979 |
|
H |
−155.007 |
−280.977 |
|
T |
−153.961 |
−280.974 |
|
V |
−155.712 |
−280.981 |
| Mg |
B |
−154.192 |
−280.987 |
|
H |
−154.038 |
−280.984 |
|
T |
−153.753 |
−280.981 |
|
V |
−154.192 |
−280.987 |
| Ca |
B |
−155.498 |
−280.987 |
|
H |
−155.511 |
−280.962 |
|
T |
−155.473 |
−280.978 |
|
V |
−155.498 |
−280.988 |
| Table 3. Optimized total energies of silicene and silicane with adatoms at different initial adsorption sites. . |
The diffusion barriers of the alkali metal atoms on silicane are extremely similar to those on silicene, about 0.20, 0.15, and 0.10 eV for Li, Na, and K respectively. In other words, hydrogenation does not change the potential surface of the silicene. Unlike the cases of alkali metal atoms, the diffusion barriers of the alkaline-earth metal atoms on silicane are significantly lower than those on silicene. Specifically, the diffusion barriers of Be, Mg, and Ca atoms on silicene are 0.91, 0.21, and 0.11 eV, whereas 0.02, 0.01, 0.01 V on silicane, respectively. Two factors result in the large difference. One is the local structure distortion after the adsorptions of alkaline-earth metal atoms on silicene, which impedes the movement of the metal atoms. This obvious distortion is not observed for the silicane substrate, thus reducing the barrier of the atom diffusion. The other factor is the weakened interaction between metal atoms and silicane, which is reflected by the adsorption energies. Weak interaction means little influence of the substrate on the movement of the adatom. Therefore, low diffusion barriers of alkaline-earth metal atoms on silicane could be understood clearly.
Considering the relatively low adsorption energies and low diffusion barriers of alkali metal atoms and alkaline-earth metal atoms on silicane, it is likely for these atoms to form clusters. In order to prove this, we calculate the cohesive energies of the different bulk metals, which are listed in Table 4. Our calculated cohesive energies of the bulk metals are very close to the experimental values from Yang et al.,[33] except for the relative large difference of Li cohesive energy, which is about 0.24 eV. According to the cohesive energies, regardless of the calculated or experimental results, the adsorption energies for all metal atoms on silicane are essentially lower than the corresponding cohesive energies of bulk metal, which indicates that the atoms prefer to gather together to form a metal cluster instead of uniformly being distributed on silicane.
Table 4.
Table 4.
Table 4. Cohesive energies of the different bulk metals. .
| Atom |
Li |
Na |
K |
Be |
Mg |
Ca |
| Eb/eV |
1.872 |
1.280 |
1.007 |
3.653 |
1.486 |
1.898 |
| Table 4. Cohesive energies of the different bulk metals. . |
Actually, the results mentioned above correspond to the adsorption and diffusion of metal atoms on perfect silicane. If the silicane substrate is defective, the situations are significantly different. According to the study reported by Yang et al.,[33] the adsorption energies of Li, Na, and K atoms on silicane with hydrogen atoms replaced by adatoms are 2.507, 2.360, and 2.623 eV, respectively. These large adsorption energies ensure that Li, Na, and K atoms could be directly adsorbed on the silicane instead of forming metal clusters. Therefore, if we need to distribute the atoms uniformly on silicane, the hydrogen vacancies should be produced in advance. As for the adsorptions of Mg and Ca atom, only the adsorption energy of the Ca atom on silicane with a hydrogen atom replaced by an adatom is slightly larger than the corresponding cohesive energy of bulk metal. As a consequence, the alkaline-earth metal atoms are not suitable for the uniform distribution on silicane, regardless of perfect or defective silicane substrate.



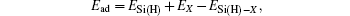



{kind=link}
{kind=link}
{kind=link}
{kind=link}
 , Lu Huan-Sheng, Liu Bo, Liu Gang, Wu Mu-Sheng, Ouyang Chuying]
, Lu Huan-Sheng, Liu Bo, Liu Gang, Wu Mu-Sheng, Ouyang Chuying]