1. IntroductionZnO has wide potential applications in ultra-violet optoelectronic devices mainly because of its band gap of 3.37 eV and exciton binding energy of 60 meV.[1–3] Fabrication of p-type ZnO, however, is still a main obstacle in achieving ZnO-based electroluminescence devices. Previous studies have shown that nitrogen as a doping element can produce shallow acceptor energy level and thus become a promising dopant for high hole concentration, high stability and reproducible p-type ZnO.[4,5] Direct doping of nitrogen into ZnO thin film is found to be very difficult in non-equilibrium growing experiments performed by most researchers. Co-doping is an effective way to solve this problem. Lithium, magnesium, gallium or other elements have been doped into ZnO together with nitrogen to acquire a more intense interaction between nitrogen and the lattice.[6–10] As isoelectronic cations in ZnO, beryllium and Mg have also attracted attention.[11] Wei et al. fabricated MgZnO:N by molecular beam epitaxy (MBE) and detected p-type conductivity after annealing.[12] Sanmyo et al. succeeded in fabricating Be–N co-doped ZnO with weak p-type conductivity by sputtering.[13] These results suggest the nitrogen-stabilizing effect of Mg and Be, which are not in agreement with calculations.[14,15] Comparison between the two neighboring group IIA dopants in ZnO helps to reveal their nitrogen-stabilizing ability, and provides guidance for further p-type doping. Differences in fabricating method and condition impede the systematic comparison between previous reports. Here we use molecular beam epitaxy, a clean growing method to avoid other disturbance, to fabricate MgZnO and BeZnO thin films under the same condition. By comparing our samples, we find that Mg and Be can both greatly enhance the stability of nitrogen in ZnO, while the nitrogen in BeZnO:N thin film stays stable even after high-temperature annealing. The changes in binding energies of Zn 2p and O 1s electrons imply an increase of oxygen content in the lattice. Luminescence from a donor–acceptor pair (DAP) is observed in BeZnO thin film. We deduce that the change in anion/cation ratio by doping affects the chemical potential of nitrogen and leads to the enhanced nitrogen stability. By comparing our results, we find that Be is better than Mg in nitrogen-stabilization. BeZnO would be a promising starting material for p-type doping.
2. Sample fabricationThe thin crystal films were grown on sapphire (0001) substrates by plasma-assisted MBE. Zn (6N), Be (4N) and Mg (6N) were supplied by solid source effusion cells. The O and N atoms were supplied from O2 and N2 gas through RF plasma generators. A designed buffer was employed to relieve the strain as much as possible and thus high crystal quality was achieved. The details of the buffer technique can be found in our previous work.[16] The doping content was controlled by the source temperature, i.e., higher source temperature corresponds to a higher doping level. In the nitrogen-doped samples, the flow rate was kept constant. The MgZnO:N thin films are n-type or highly resistant. The BeZnO:N thin films are highly resistant or weakly p-type, with a hole concentration of ∼ 1 × 1015/cm3 and a mobility of 2 cm2·V−1·S−1. The electrical properties of the films are stable at least for three months.
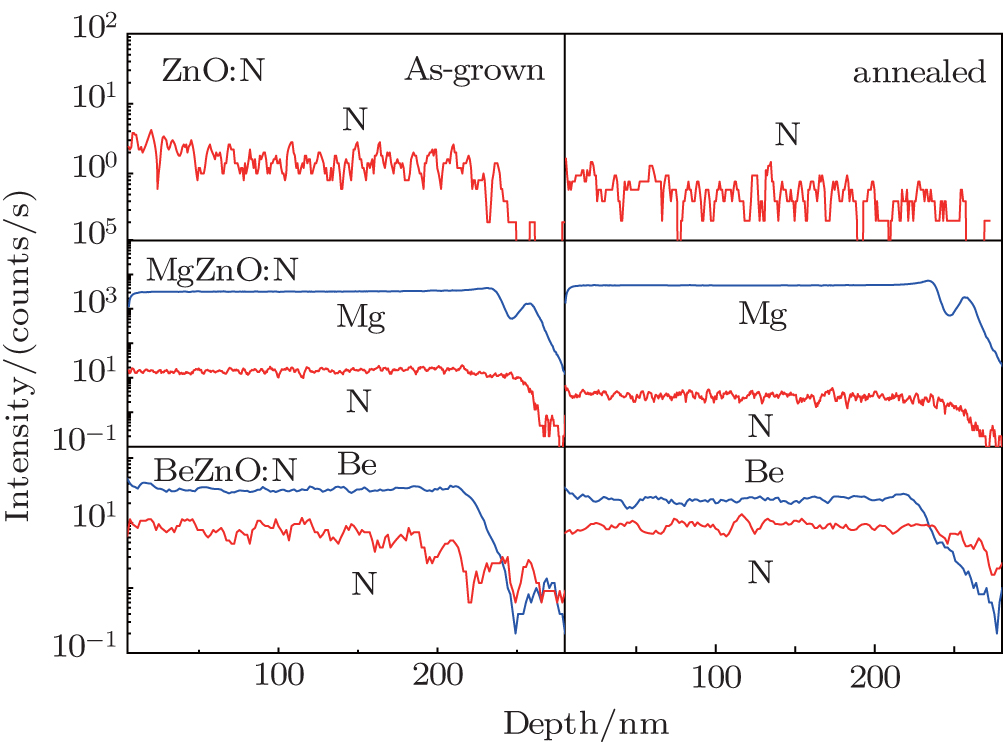
3. Experimental results and discussion3.1. Secondary ion mass spectrum measurementSecondary ion mass spectrum (SIMS) is used to investigate the relative nitrogen density in as-grown and annealed ZnO, BeZnO, and MgZnO films. Figure 1 shows the SIMS results of the nitrogen-doped films before and after annealing at 500 °C in N2 atmosphere. Nitrogen behaves differently in three samples. In the pure ZnO, the nitrogen counts are rather low even before annealing. In the MgZnO alloy, the nitrogen signal is strong in the as-grown sample but declines after annealing. In the BeZnO alloy, the nitrogen signal, although not as intense as in MgZnO for as-grown samples, becomes stronger and more uniform after annealing. Actually, the annealed BeZnO alloy has a nitrogen content value comparable to the annealed MgZnO alloy, both in concentration and uniformity. Moreover, as an experimental fact, MgZnO takes the nitrogen-stabilizing effect only for an Mg content value larger than ∼ 10% while BeZnO can readily show the effect for a Be content value less than ∼ 2%. Be as a co-doping element may be more powerful in stabilizing N dopant. These can be in part explained from the aspect of bond energy. The standard formation enthalpy for Zn3N2 is −20 kJ·mol−1, which is much smaller than that for ZnO (−348.28 kJ·mol−1).[17] This leads to the poor solubility of nitrogen in ZnO. On the other hand, the formation enthalpy values for Mg3N2[18] and Be3N2[19] are −461.08 kJ·mol−1 and −570 kJ·mol−1, respectively. As a result, Mg and Be can help to ameliorate the stability of nitrogen in ZnO. Our SIMS measurement demonstrates that nitrogen prefers MgZnO than BeZnO in as-grown samples, while BeZnO:N is stabler than MgZnO:N after annealing. This is due to the stronger Be–N bond.
The high concentration of nitrogen in the as-grown MgZnO implies that nitrogen can be easily doped into the lattice during growth. But the nitrogen losses when the system process proceeds towards thermal equilibrium state. These results agree with those given by Gai et al., who predicted a higher formation energy of neutral defect NO in MgZnO than in ZnO, but succeeded to fabricate MgZnO:N by modifying chemical potentials during nonequilibrium growth.[6] On the other hand, nitrogen in BeZnO seems to be very stable even under thermal equilibrium state. However, this experimental fact is somewhat against the calculation which predicted that incorporating Be into ZnO:N can lower the ionization energy but disadvantage the stability of nitrogen.[15] To clarify the origin of this divergence, we reconsider the equation of formation energy used in calculation. The formation energy of neutral defect NO in ZnO is given by
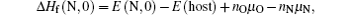
where
E(N,0) is the total energy of the supercell with neutral N
O,
E (host) is the energy of the original supercell,
μO and
μN are the chemical potentials of constituents O and N relative to the element gas energy,
nO and
nN are the numbers of O and N. The system is less stable with higher formation energy. Calculations indicate that (
E(N,0)–
E(host)) is higher in MgZnO
[6] and BeZnO
[15] than in pure ZnO. That induces a lower formation energy of N
O in ZnO than in BeZnO and MgZnO, which is in contradiction with our experimental results. Therefore, the chemical potential, which is kept constant in most calculations, needs to be considered carefully.
The chemical potential is strongly affected by stoichiometry of the lattice. Our previous work indicates that the anion/cation ratio in BeZnO alloy may change from 1 to over 1.1 by increasing Be doping. The positron annihilation spectrum also evidences that the oxygen vacancies are obviously suppressed by Be doping.[20] But no data have ever addressed that point for MgZnO. Here, on that point, we provide more detailed spectrum evidence for both BeZnO and MgZnO: photoluminescence (PL) and x-ray photoelectron spectrum (XPS) measurements are carried out. We compare samples with and without nitrogen doping to exclude the influence of nitrogen. It is found that no change is caused by nitrogen doping in the spectrum region we are interested in. This is because the nitrogen concentration is rather low compared with those of Be and Mg.
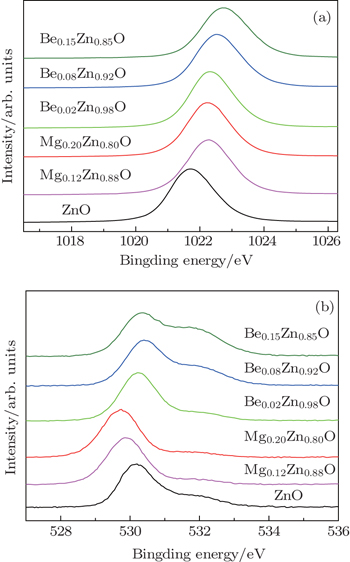
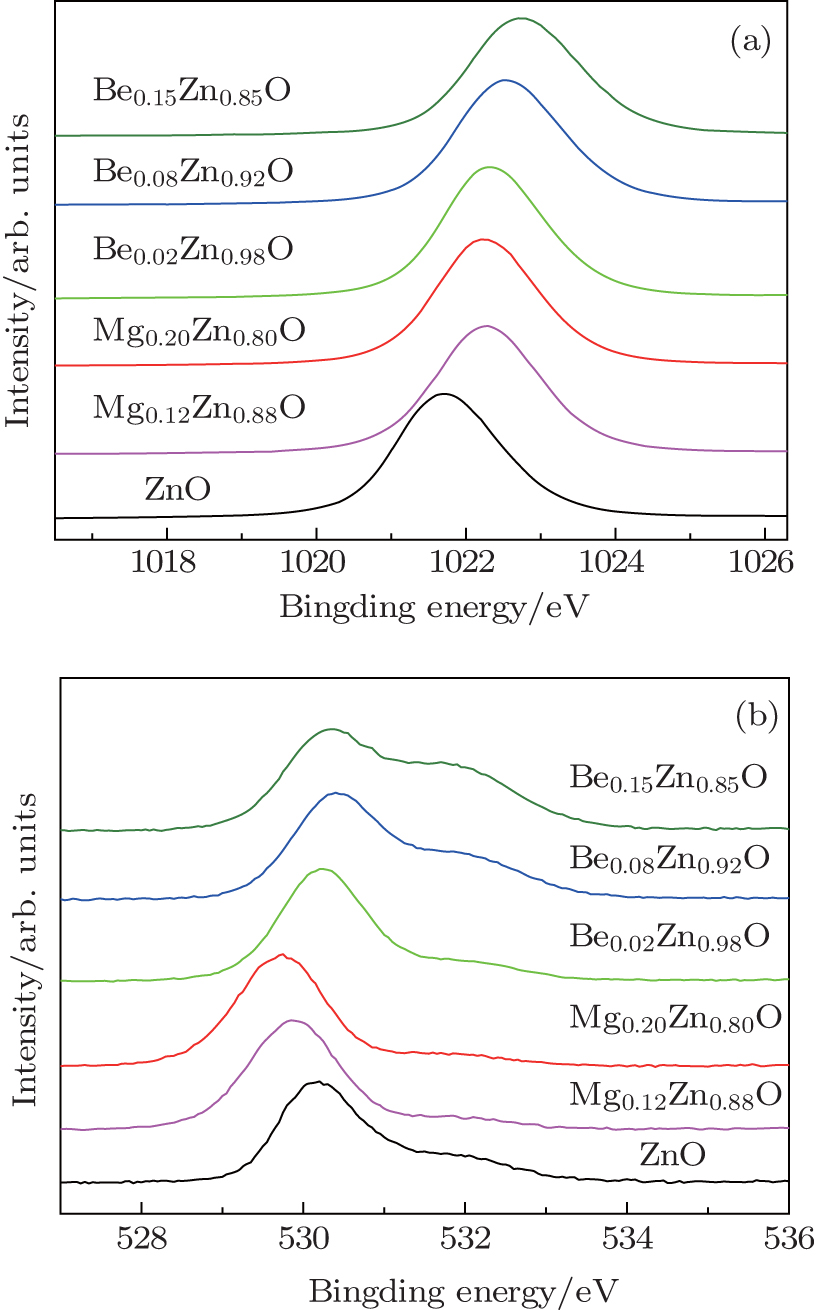
3.2. X-ray photoelectron spectrumFigure 2(a) shows XPS Zn 2p3/2 binding energy spectra from ZnO, MgZnO, and BeZnO. Both dopants can increase the binding energies. For Mg doping, the peak shifts to higher energy in Mg0.12Zn0.88O and then shifts back in Mg0.2Zn0.8O. The shift for BeZnO with a wide range of Be content is larger than for MgZnO. The variation of the Zn 2p binding energy is related to the ionicity of ZnO.[21,22] When zinc atoms are bonded to oxygen, its core level binding energy will increase. It means the binding energy of Zn 2p electron should increase with increasing oxygen content. Therefore, the ratio of oxygen atoms to metal atoms is increased by Mg or Be doping, and Be seems to be more efficient. This agrees with the oxygen content obtained from XPS in Table 1. In ZnO, oxygen vacancies are abundant and the crystal is not stoichiometric, i.e., there are more Zn atoms than O atoms. This situation changes in these alloys. Oxygen vacancies are suppressed by Mg and Be. Note that the PL and XPS results mentioned above are observed in BeZnO and MgZnO in the case without nitrogen doping. The advantages brought by Be or Mg doping may also be beneficial when using acceptor dopants other than nitrogen.
Table 1.
Table 1.
 Table 1. Oxygen content of ZnO films doped with different content values. .
| Material |
ZnO |
Mg0.12Zn0.88O |
Mg0.2Zn0.8O |
Be0.02Zn0.98O |
Be0.08Zn0.92O |
Be0.15Zn0.85O |
|---|
| Oxygen content |
45% |
47% |
47% |
47% |
48% |
53% |
| Table 1. Oxygen content of ZnO films doped with different content values. . |
Modification of the O 1s binding energy by doping is also observed. As shown in Fig. 2(b), the O 1s peak is composed of at least two components. The main component located at approximately 530 eV originates from oxygen at the ideal lattice site. It shifts slightly with doping due to the difference among the 1s bonding energies of oxygen bonded to Zn, Mg, and Be. The shoulder located at about 532 eV has been reported in ZnO[23,24] and other oxides.[25,26] It originates from oxygen atoms near the deficient region of the lattice. The shoulder peaks are weak in ZnO and MgZnO but become remarkable in BeZnO. This shoulder peak should relate to oxygen atoms near the acceptor defects in our study.
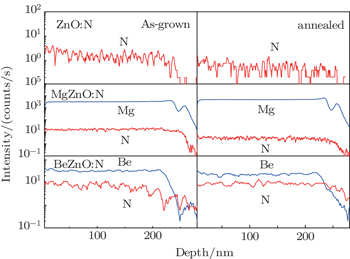
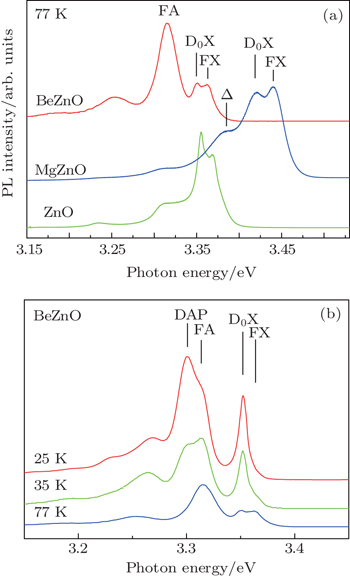
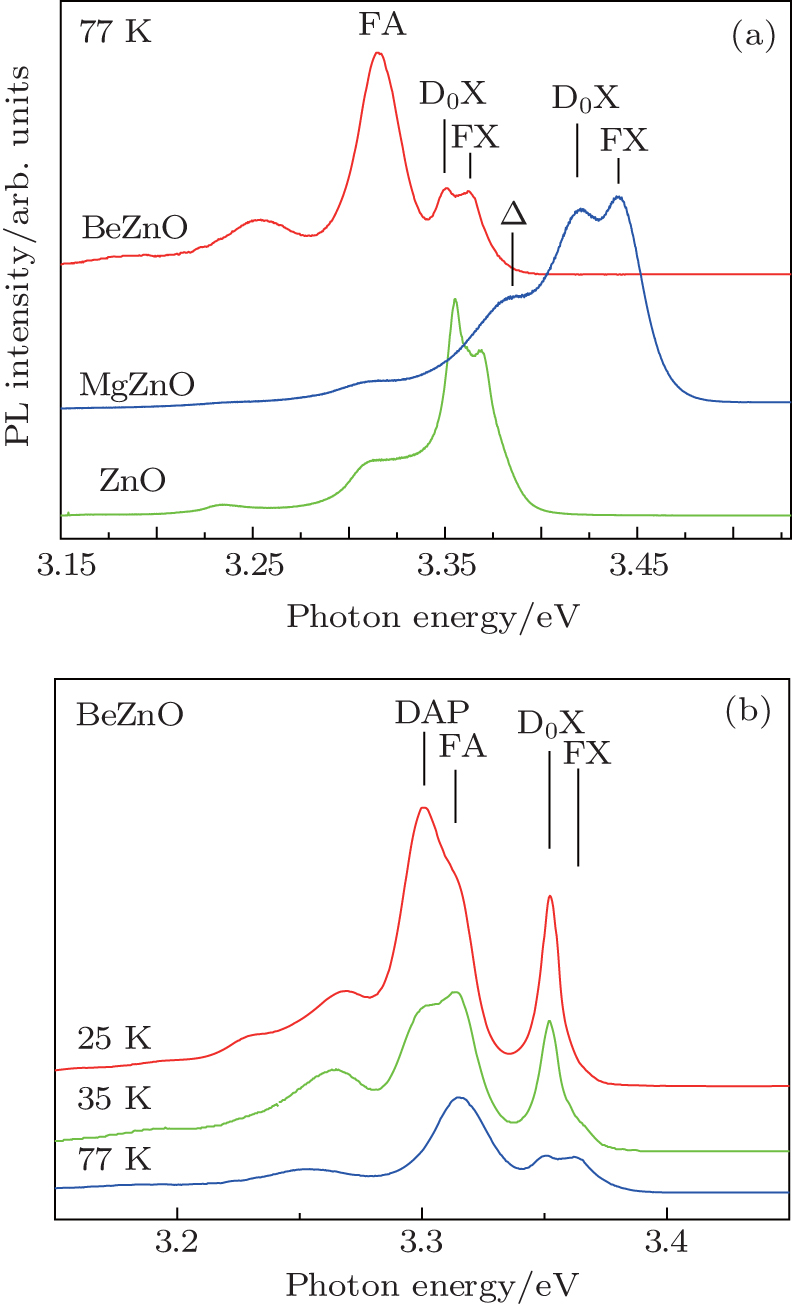
3.3. Photoluminescence spectrumThe PL spectra of ZnO, MgZnO, and BeZnO are shown in Fig. 3(a). Only luminescences from free exciton (FX), exciton bonded to neutral donor (D0X) and their replicas are observed in ZnO. In MgZnO, the broad band on the low energy side of D0X (denoted as Δ) becomes stronger in intensity. This implies the existence of a new peak such as luminescence from the donor–acceptor pair (DAP), which was also observed by Li et al.[27] As this peak overlaps with the replicas of D0X and FA, it is difficult to locate it precisely. This is the case for a wide range of Mg content (judging from the enlargement of band gap). In the BeZnO sample, the band gap is barely affected since the Be content is kept low (2%). But an intense new peak appears at 3.316 eV. To investigate the origin of this peak, we measure the temperature-dependent PL of BeZnO (Fig. 3(b)). When the temperature decreases from 77 K to 25 K, another peak appears at ∼ 3.3 eV and becomes dominant gradually. The position of the 3.3 eV peak blue-shifts with increasing laser power (not shown in the figure). We attribute this peak to the DAP luminescence peak. The energy of the photon resulting from radioactive recombination of DAP is given by[28]
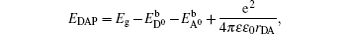
where
Eg is the band gap energy, are the binding energies of neutral donor and acceptor, respectively,
rDA is the distance between the donor and the acceptor in the lattice. The DAP luminescence is contributed by the recombination of DAPs with different
rDA values and thus different energies. When the exciting power increases, contributions from DAPs with smaller
rDA values increase. Therefore, the peak blue-shifts. At higher temperature, the DAP recombination is quenched and the peak is replaced by luminescence from the transition of free electrons to acceptors (FA). This temperature-dependent evolution is a typical feature of DAP. The BeZnO sample used here is not intentionally doped with other elements, so the acceptor is attributed to being V
Zn.
[27] ZnO always presents a dense n-type background. The existence of this acceptor may help to compensate for this.
3.4. Further discussionAll the above evidence points to the same conclusion: the anion/cation ratios in BeZnO and MgZnO alloys increase and the anion/cation ratio is much more remarkable in BeZnO. This conclusion may be a way out for the problematic calculations mentioned above, because most of the calculated values of (E(N,0)–E(host)) are obtained on an assumed lattice with anion/cation ratio r = 1, which is no longer valid when the doping elements significantly affect the anion/cation ratio and break the balance of intrinsic defects. We suggest that it is more reasonable for the calculation work to take the variation of anion/cation ratio into account. The degree of the variation may take the experimental data as a reference. Whether this modification will do any favors to the model is still an open question waiting for its answer.
4. SummaryIn this paper, we fabricate high-quality MgZnO:N and BeZnO:N alloy films by MBE under a similar growth condition. The nitrogen stability is confirmed by SIMS in both alloys. To study the nitrogen-stabilizing mechanism, we examine the BeZnO and MgZnO alloys in more detail by PL and XPS. In PL spectra, an additional donor–acceptor pair irradiative emission is observed in BeZnO while in MgZnO the emission signal is not so intense. This new emission peak implies the decrease of O vacancy and (or) the increase of Zn vacancy. On the other hand, XPS indicates that the content values of oxygen are increased in both alloys, which accords well with the implication from the PL spectrum. These phenomena are all related to the doping-induced modification of host element atomic ratio, which is not considered in calculations. Also from our experiments, the N stabilization and O vacancy suppression are both more prominent in BeZnO than in MgZnO. Thus Be is a better co-doping element than Mg for p-type ZnO:N.


{kind=link}
{kind=link}
{kind=link}
 , Chen Ming-Ming2, 3, †,
, Chen Ming-Ming2, 3, †,