{kind=link}
{kind=link}
{kind=link}
Correlation between valence electronic structure and magnetic properties in RCo5 (R = rare earth) intermetallic compound
[Xue Zhi-Qin, Guo Yong-Quan†,  ]
]
]
|
|


† Corresponding author. E-mail:
Project supported by the National Natural Science Foundation of China (Grant No. 11274110).
The magnetisms of RCo5 (R = rare earth) intermetallics are systematically studied with the empirical electron theory of solids and molecules (EET). The theoretical moments and Curie temperatures agree well with experimental ones. The calculated results show strong correlations between the valence electronic structure and the magnetic properties in RCo5 intermetallic compounds. The moments of RCo5 intermetallics originate mainly from the 3d electrons of Co atoms and 4f electrons of rare earth, and the s electrons also affect the magnetic moments by the hybridization of d and s electrons. It is found that moment of Co atom at 2c site is higher than that at 3g site due to the fact that the bonding effect between R and Co is associated with an electron transformation from 3d electrons into covalence electrons. In the heavy rare-earth-based RCo5 intermetallics, the contribution to magnetic moment originates from the 3d and 4f electrons. The covalence electrons and lattice electrons also affect the Curie temperature, which is proportional to the average moment along the various bonds.
Owing to the large magnetocrystalline anisotropies, high Curie temperatures and high saturation moments at room temperature, RCo5 (R = rare earth) intermetallic compounds have received considerable scientific and technological attention in the past decades,[1,2] and the studies have covered the experiment, theory and technology for developing the advanced rare earth permanent magnetic materials.[3–5] The study on magnetic properties of RCo5 intermetallic compounds is derived from the magnetization measurements on single crystals.[6] Rao and Greedan have established the relationships between the basic parameters in crystal-field theory and the magnetocrystalline anisotropy in most RCo5 intermetallic compounds.[7] The single-ion anisotropy has been found to have a fundamental influence on the behavior of the magnetic system and the related mechanism has been studied.[8,9] In many cases, the single-ion anisotropy model contains many calculating parameters especially in the Heisenberg model proposed based on the exchange coupling interaction. As for the first principle theory, the magnetic properties of RCo5 compounds are usually discussed within a two-sublattice model consisting of R and Co sublattices.[10,11] The computational methods based on the density functional theory (DFT) provide an important insight into the electronic structure and the related physical properties for the various materials. The basic DFT approximation (local spin density approximation-LSDA) is usually restricted to the so-called open core treatment of the 4f electrons.[12,13] However, LSDA could not predict the energetics of the various magnetic orders due to the absence of the energy gap between the filled and the empty states and underestimations of the orbital magnetic moments.[14,15] The theoretical work on the anisotropy and Curie temperature of R–(Fe,Co) magnetic alloys were carried out in the molecular-field approximation by studying the exchange coupling interactions between the 3d electrons of Fe or Co and 4f electrons of rare earth.[16] However, there are too many theoretical parameters in the complicated calculation. Also, the samples should be of the single-crystal.[17]
As for the first principle theory, it could not initially be used to study the magnetism of rare-earth-based intermetallic compounds. In recent years, a framework of the LSDA + U approach has been developed to calculate the magnetic properties of rare earth alloys and compounds based on the mean-field theory.[18] However, the calculation is restricted by the structural cell model and the capacity of the computer. In the present paper, we use the empirical electron theory of solids and molecules (EET), which is based on Pauling’s electron theory of metals.[19] Owing to its lesser computing parameters than the molecular-field and the first principle theories, the model is simpler than the theories of crystal-field and first-principle without considering any integral or differential calculation. The EET can be used to investigate the correlations between the valence electronic structures and multiple physical properties of solid state materials. The approximations, which are based on the electronic structures, can work in studying the essential mechanisms of physical phenomena as reported. On one hand, the electronic structure analyses could reveal many natures of physical phenomena such as: orbital hybridization, band gap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), vertical ionization potentials, and electron affinities, which are important for studying the correlations between the electronic structures and physical properties in molecules and solids.[20–22] The multiple properties such as cohesive energy, melting point, transition temperature, magnetic moment, Curie temperature, superconductor temperature, etc. have been calculated based on the electron states in the electronic structure.[23–26] In this study, the magnetic moments and Curie temperatures of RCo5 (R = Y, La, Ce, Pr, Nd, Sm, Gd, Tb, Dy, Er) intermetallics are systematically studied with EET and the relations between the electronic structures and magnetic characteristics of these compounds are discussed as well.
EET is a semi-empirical theory based on the Pauling valence-bonding theory and Hume–Rothery electron concentration rule. As a supplement, the equivalence valence electron hypothesis is introduced for the application of the electron theory to the crystal structures. The theory consists of three general hypotheses and one method of calculating bond distances. Hypothesis 1: the atoms in a solid and a molecule are in general hybridization of the two states, which can be called the h (head) and t (tail) states. Both states have one or two kinds of valence electrons; that is, covalent and newly introduced lattice electrons. Hypothesis 2: the hybridization of the two states is discontinuous, allowing only certain relative compositions to be calculated by a k-equation. Hypothesis 3: the covalent bond length between atom u and v with the number of covalent electron pairs denoted by nα is related by
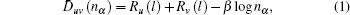
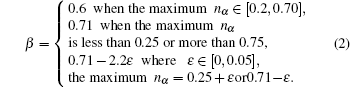
From the derivation, it is seen that
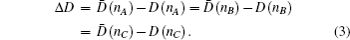
According to the criterion of ΔD ≤ 0.05 in the BLD method, the hybridization states of the electrons in solid and molecules could be determined, and the relative values of Chσ and Ctσ are determined.
Then there is given the equation for calculating the magnetic moments:
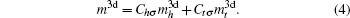
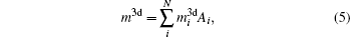
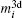
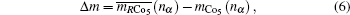

The magnetic moments of rare earth 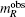
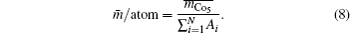
The model for calculating Curie temperature is based on the assumption of molten ferromagnetic order structure by the heat vibrations induced by the phonon energy with increasing temperature. When the temperature reaches a certain value of Tc, the phonon energy equals the magnetic energy. If T > Tc, the magnetic order is molten and the ferromagnetic characteristics disappear. This transition temperature is regarded as the Curie temperature. The energy of N phonons is
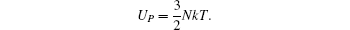
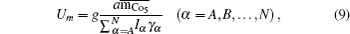
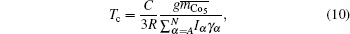
Then here is given the criterion, ΔTc.
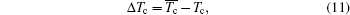
In summary, the theoretical
| Table 1. Lattice parameters of RCo5 and valence bond lengths. . |
| Table 2. Calculation results of magnetic properties. In this table, nα (max) denotes the maximum number in the covalent electron pairs nα (α = A,B, … N), and the symbol “–” refers to the negative moment originating from the heavy rare earths since their moment arrangements are anti parallel to the direction of 3d moment of Co. . |
| Table 3. Analyses of valence electronic structures of RCo5. In this table, nc, nl, m3d, and nT denote the numbers of the covalent electrons, the lattice electrons, the magnetic electrons, and the total number of the electrons, respectively; σ is the number of quantum states. The rare earth element has a piece of the hybridization table, which possesses 10 kinds of quantum states. Element Co has 8 pieces of the hybridization table (A1, A2, B, C1, C2, D, E1, E2), and each of them records 18 kinds of quantum states. . |
RCo5 compounds crystallize into the hexagonal CaCu5-type structure with a space group of P6/mmm, and the atomic occupations are 1a for R, 2c for CoI, 3g for CoII, respectively. The lattice parameters of RCo5 compounds and the valence bond lengths of R–Co and Co–Co bonds are listed in Table
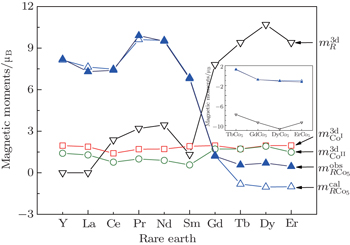
The calculated results of magnetic properties are given in Table 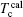
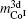
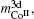
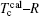
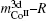
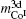
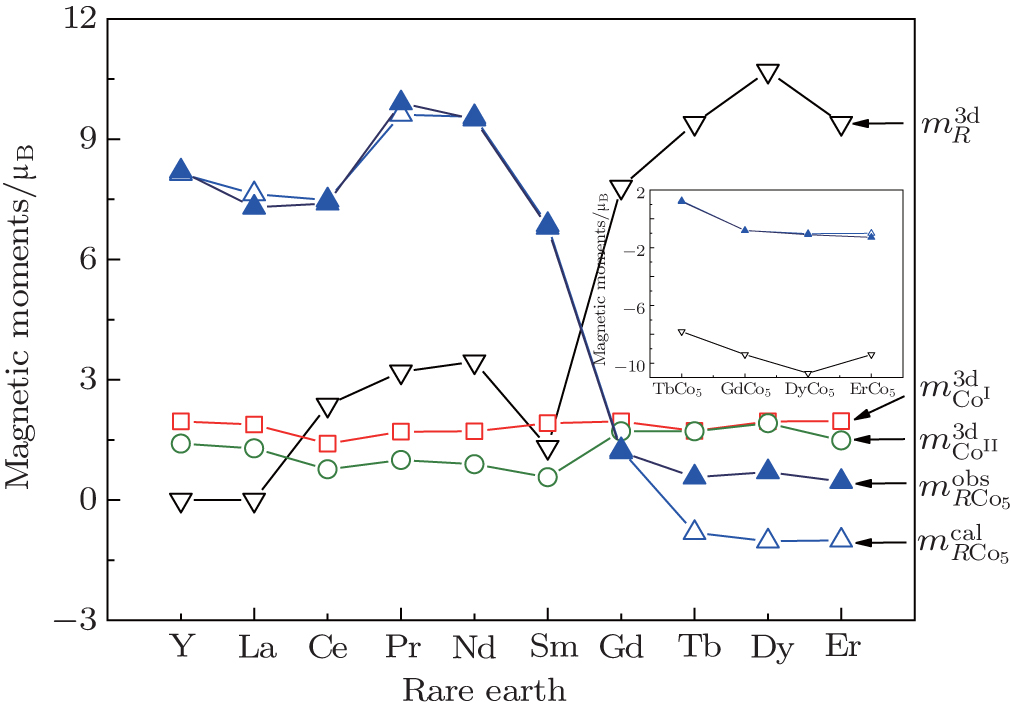 | Fig. 1. Calculated results of magnetic moments of R-, 2c- and 3g-sited Co in RCo5 compounds. The insert gives the antiferromagnetic interaction between R and Co in RCo5 compounds when R = heavy rare earth. |
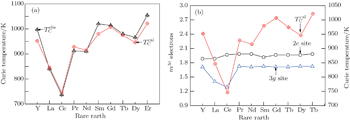
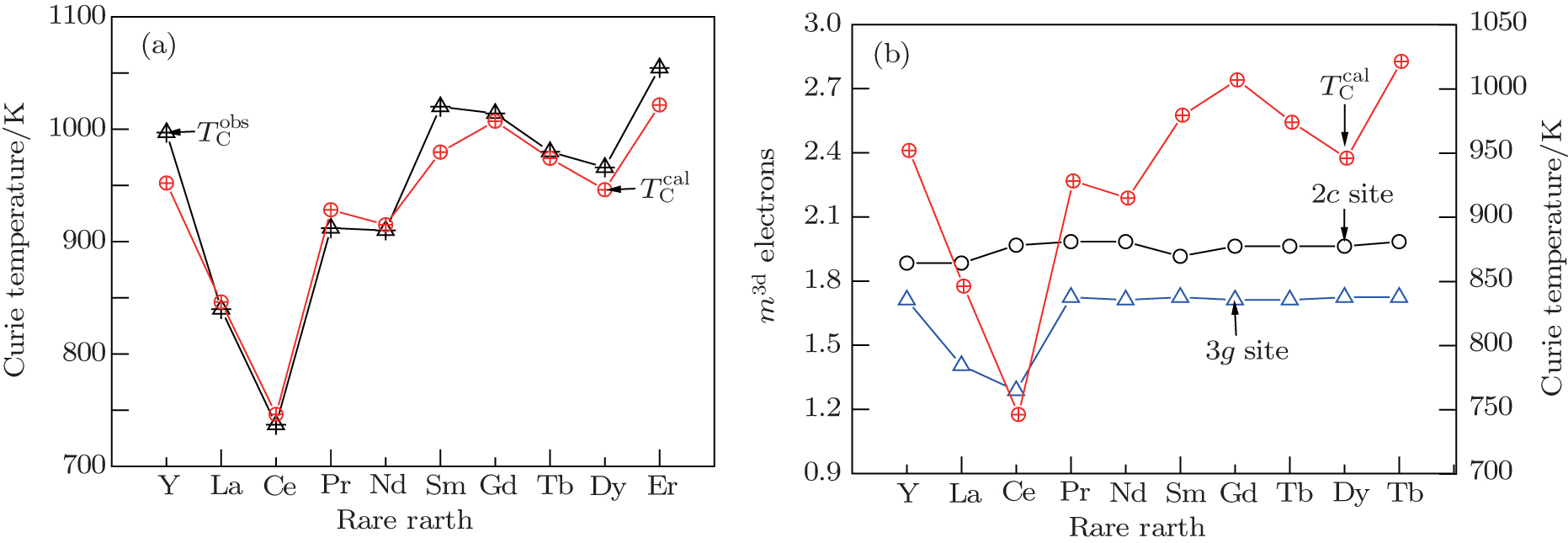 | Fig. 2. (a) Observed and calculated Curie temperatures of RCo5 and (3d) electron numbers of Co atoms at 2c and 3g sites. |
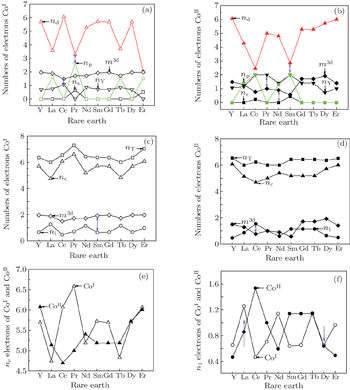
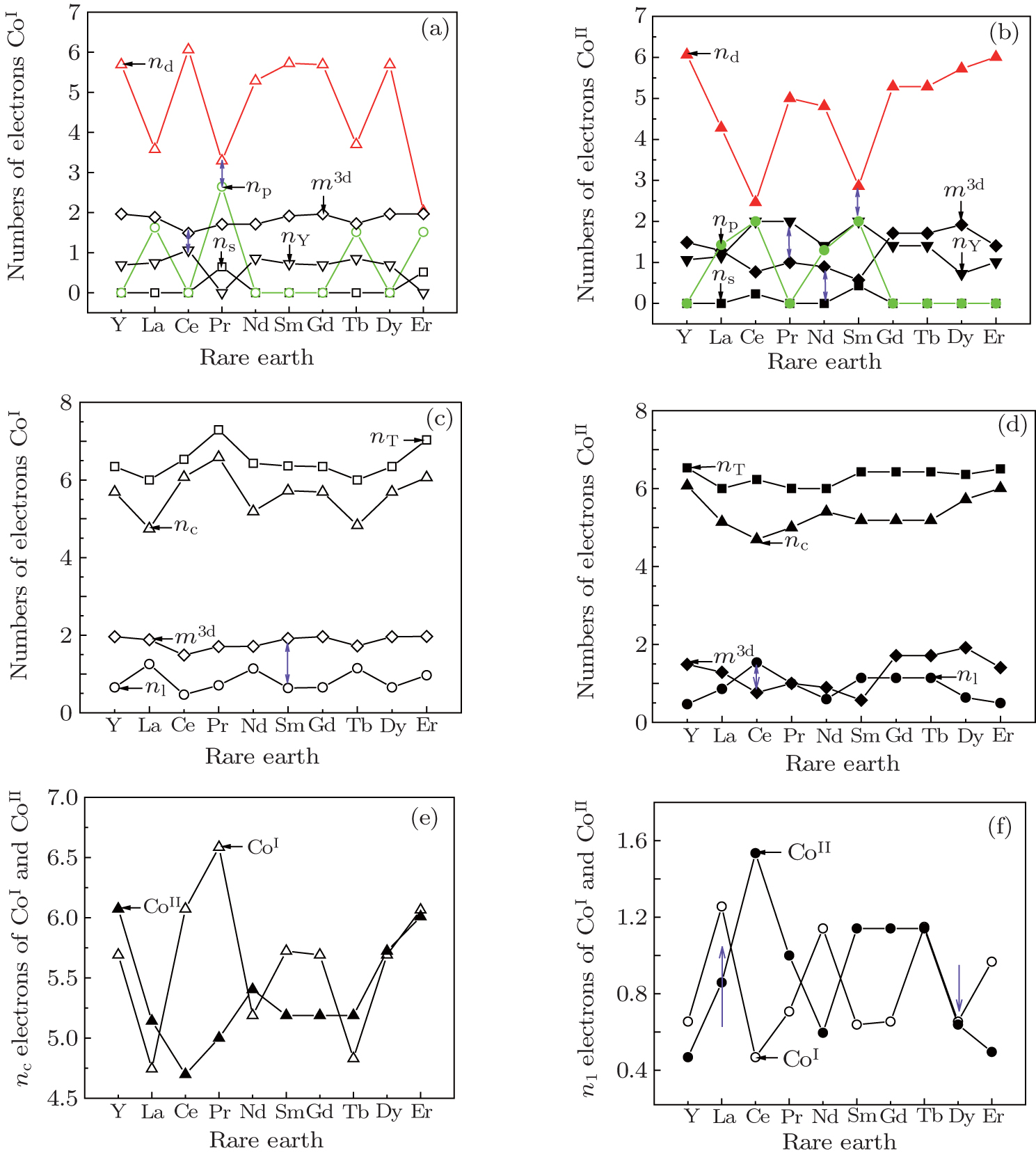 | Fig. 3. Electronic transformations among all kinds of electrons. The electronic distributions of Co at 2c site (a) and 3g site (b); the electron transformations among the covalence, lattice, 3d magnetic electrons are shown for Co at 2c site (c) and for Co at 3g site (d); the covalence and lattice electrons distributions for CoI and CoII are shown in panels (e) and (f). |
The calculated valence electronic structures are given in Table
Figures
The empirical electron theory of solids and molecules (EET), which is based on the Pauling’s valance theory, is used to investigate the valence electronic structures and multiple physical properties of solid state materials. The theoretical model is simpler with fewer theoretical parameters than the first principle and the molecular field theory. In this study, the magnetic moments and Curie temperatures of RCo5 (R = rare earth) intermetallics are systematically studied with EET.
The calculated results reveal that the moments of RCo5 intermetallics originate mainly from the 3d electrons of Co atoms and 4f electrons of rare earth, the s electrons also affect the magnetic moments by the hybridization of d and s electrons. The contribution to the moment depends on the sublattices of Co and R atoms, and it is found that the moment of Co atom at 2c site is bigger than that at 3g site due to the bonding effect between R and Co associated with a electron transformation from 3d electrons into covalence electrons. In the heavy rare-earth-based RCo5 intermetallics, the contribution to the molecular moment originates from 3d and 4f electrons. Curie temperatures of RCo5 are proportional to the average moment along the various bonds, and it indicates the magnetic anisotropy along the various crystal directions. The covalence electrons and lattice electrons also affect the Curie temperature. RCo5 intermetallic compounds show strong relationships between their valence electronic structure and magnetic properties. Except for the 3d–3d or 3d–4f exchange coupling interaction contributing to the ferromagnetic order, the lattice electrons also affect the Curie temperature.
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 | |
| 22 | |
| 23 | |
| 24 | |
| 25 | |
| 26 | |
| 27 | |
| 28 |