








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
First-principles study of the elastic and thermodynamic properties of thorium hydrides at high pressure
[Zhang Xiao-Lin1, Wu Yuan-Yuan1, Shao Xiao-Hong1, †,  , Lu Yong2, Zhang Ping2, ‡, ]
, Lu Yong2, Zhang Ping2, ‡, ]
, Lu Yong2, Zhang Ping2, ‡, ]
|
|


† Corresponding author. E-mail:
‡ Corresponding author. E-mail:
Project supported by the Long-Term Subsidy Mechanism from the Ministry of Finance and the Ministry of Education of China.
The high pressure behaviors of Th4H15 and ThH2 are investigated by using the first-principles calculations based on the density functional theory (DFT). From the energy–volume relations, the bct phase of ThH2 is more stable than the fcc phase at ambient conditions. At high pressure, the bct ThH2 and bcc Th4H15 phases are more brittle than they are at ambient pressure from the calculated elastic constants and the Poisson ratio. The thermodynamic stability of the bct phase ThH2 is determined from the calculated phonon dispersion. In the pressure domain of interest, the phonon dispersions of bcc Th4H15 and bct ThH2 are positive, indicating the dynamical stability of these two phases, while the fcc ThH2 is unstable. The thermodynamic properties including the lattice vibration energy, entropy, and specific heat are predicted for these stable phases. The vibrational free energy decreases with the increase of the temperature, and the entropy and the heat capacity are proportional to the temperature and inversely proportional to the pressure. As the pressure increases, the resistance to the external pressure is strengthened for Th4H15 and ThH2.
Thorium and its hydrides are promising materials for advanced nuclear reactors with a high level of safety[1–8] and have attracted much attention. This is mainly due to their strength and stability at elevated temperatures while maintaining the desirable properties for neutron moderation.[9] In general, thorium hydrides crystalize in ThH2 and Th4H15 phases, which are both metallic solids.[2] Under ambient conditions, the stable thorium dihydride crystallizes in a body-centered tetragonal (bct) ionic structure with space group I4/mmm (No. 139). There also exists a face-centered cubic (fcc) fluorite-type structure with space group Fm-3m (No. 225), which is considered as a metastable phase of ThH2. As for the Th4H15 phase, it crystallizes in a body-centered cubic (bcc) structure with space group I-43d (No. 220), which has been reported to have superconductivity with a transition temperature of 8 K.[10] In addition, due to the large hydrogen-to-metal ratio, Th4H15 has been considered as a promising candidate for hydrogen storage.[11]
The structural and electronic properties and the optical phonon density of states of thorium hydrides at the ground state have been widely investigated in previous studies.[12–19] In order to support the experimental measurements or predict the microscopic physical properties that cannot be directly measured in the experiments, many theoretical studies were also performed for thorium hydrides. The optical phonon density of states of ThH2 was measured through inelastic neutron scattering,[14] and the bulk modulus of ThH2 was calculated by the linear muffin-tin orbital (LMTO) method.[15] The electronic properties of thorium hydrides were investigated by Shein et al.[16] through the full-potential LAPW (FLAPW) method. Moreover, the structural, elastic, and electronic properties of thorium hydrides at the ground state were studied by the first-principles calculations.[17] Despite abundant researches on thorium hydrides, relatively little is known regarding Th4H15. Until now, the elastic properties, phonon spectra, and thermodynamic properties are still unknown for Th4H15. Besides, there is still a lack of data about the elastic and thermal properties of thorium hydrides under high pressures, where a variety of interesting changes could show up. Furthermore, the stability of such competitive phases of thorium hydrides is also of interest for testing their practical applications.
In this paper, we perform a first-principles study on the elastic and thermodynamic properties of thorium hydrides at high pressures and make a systematic comparison with the corresponding behaviors at the ground state. The calculation method for the elastic constants and the computational details of first-principles are briefly introduced in Section 2. The calculation results are presented and discussed in Section 3. Finally, the conclusion is given in Section 4.
The first-principles calculations were carried out by using the density-functional theory (DFT) as implemented in Vienna ab initio simulations package (VASP)[20] with the projector-augmented-wave (PAW) potential methods.[21] The electron exchange and correlation potential was evaluated by the generalized gradient approximation (GGA) introduced by Perdew, Burke, and Ernzerhof (PBE).[22] To ensure the calculation accuracy, the cutoff energy was set to 450 eV, and the self-consistent energy convergence of the calculation was set to 10−5 eV. The Brillouin zone (BZ) integration was performed by using 12 × 12 × 12k-points for ThH2 and 9 × 9 × 9 k-points for Th4H15 with the Monkhorst–Pack scheme.[23] The atomic structures were fully relaxed until the Hellmann–Feynman forces on all atoms were less than 1.0 × 10−5 eV/atom.
The elastic constants were calculated by means of a Taylor expansion of the total energy E(V,δ) based on the following law:[24–26]

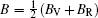
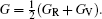
The Helmholtz free energy F (T,V) was calculated by





In the present study, we investigated three phases of thorium hydrides, i.e., bct ThH2, fcc ThH2, and bcc Th4H15(Fig.
 | Fig. 1. Crystal structures of (a) bct ThH2, (b) fcc ThH2, and (c) bcc Th4H15. The larger green balls are Th atoms and the smaller white ones are H atoms. |
 | Fig. 2. Calculated ground-state energy vs. the primitive cell volume for ThH2 in bct and fcc phases. |
Elastic constants are important mechanical parameters of solid materials, which determine the stiffness of a crystal against the external strain. Thus, we calculate the elastic constants to study the mechanical properties of thorium hydrides under different pressures. The calculated lattice parameters, elastic constants Cij, bulk modulus B, shear modulus G, Young’s modulus E, and Poisson’s ratio ν for bct ThH2, fcc ThH2, and bcc Th4H15 at different pressures are listed in Table
| Table 1. Lattice parameter, elastic constants Cij, bulk modulus B, shear modulus G, Young’s modulus E, and Poisson’s ratio ν of bct ThH2 at different pressures. . |
| Table 2. Lattice parameter, elastic constants Cij, bulk modulus B of fcc ThH2 at different pressures. . |
| Table 3. Lattice parameter, elastic constants Cij, bulk modulus B, shear modulus G, Young’s modulus E, and Poisson’s ratio ν of bcc Th4H15 at different pressures. . |
As for the cubic structure, the structural stability criteria are C11 > 0, C44 > 0, C11 > |C12|, (C11 + 2C12) > 0. From the criteria, it is found that the bcc Th4H15 is mechanically stable, while the fcc ThH2 is unstable since C11 − C12 is negative. Nearly all the elastic constants increase with increasing pressure except C12 and C13 for the bct phase and C11 for the fcc phase (Fig.
 | Fig. 3. Calculated elastic constants as a function of pressure for (a) bct ThH2, (b) fcc ThH2, and (c) bcc Th4H15. |
The increasing rates of bulk moduli in these phases are slightly lower than those of shear moduli. It is well known that a high ratio of B/G is responsible for the ductility of polycrystalline materials. The critical value which separates ductile and brittle materials is around 1.75.[32] Our calculated B/G decreases from 3.29 to 1.37 for the bct phase with the pressure increasing from 0 to 60 GPa (Fig.
 | Fig. 4. Dependencies of B/G ratio on pressure for (a) bct ThH2 and (b) bcc Th4H15. |
In this work, the phonon dispersion was calculated based on the supercell approach and the finite difference method as implemented in the PHONOPY code. The calculated phonon dispersions for bct ThH2, fcc ThH2, and bcc Th4H15 are shown in Fig. 

 | Fig. 5. Calculated phonon dispersions (left panel) and phonon DOS (right panel) for (a) bct ThH2, (b) fcc ThH2, and (c) bcc Th4H15. |
 | Fig. 6. Thermodynamic properties of (a) bct ThH2 and (b) bcc Th4H15 as a function of temperature under various pressures. |
 | Fig. 7. Dependences of the Helmholtz free energy F(T,V) on crystal volume at various temperatures for (a) bct ThH2 and (b) bcc Th4H15. |
 | Fig. 8. Temperature dependences of bulk modulus B(T) of (a) bct ThH2 and (b) bcc Th4H15. |
 | Fig. 9. Specific heat capacities of (a) bct ThH2 and (b) bcc Th4H15. |
The high pressure behaviors of Th4H15 and ThH2 have been studied by using the first-principles calculations based on the density functional theory. From the energy–volume relationship, the bct phase of ThH2 is more stable than the fcc phase at ambient pressure. At high pressures, the bct ThH2 and bcc Th4H15 phases are more brittle than they are at ambient pressure in terms of the calculated elastic constants and the Poisson ratio. The thermodynamic stability of these three phases has been discussed with the calculated phonon dispersion curves. In the pressure domain of interest, the phonon dispersions of bcc Th4H15 and bct ThH2 are positive, indicating that these two phases are stable, while the fcc ThH2 is unstable. The thermodynamic properties including the lattice vibration energy, entropy, and specific heat have been predicted based on the vibrational frequencies for the stable phases. Our results show that the vibrational free energy decreases with the increase of the temperature, while the entropy and the heat capacity are proportional to the temperature and inversely proportional to the pressure. It is found that the bulk modulus B increases with pressure, indicating the strengthened resistance to the external pressure for Th4H15 and ThH2. The calculated results in this study would be helpful for future experimental measurements on thorium hydrides.
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 | |
| 22 | |
| 23 | |
| 24 | |
| 25 | |
| 26 | |
| 27 | |
| 28 | |
| 29 | |
| 30 | |
| 31 | |
| 32 | |
| 33 | |
| 34 |