






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of pressure on electronic and thermoelectric properties of magnesium silicide: A density functional theory study
[Kaur Kulwinder†,  , Kumar Ranjan]
, Kumar Ranjan]
, Kumar Ranjan]
|
|


† Corresponding author. E-mail:
Project supported by the Council of Scientific & Industrial Research (CSIR), India.
We study the effect of pressure on electronic and thermoelectric properties of Mg2Si using the density functional theory and Boltzmann transport equations. The variation of lattice constant, band gap, bulk modulus with pressure is also analyzed. Further, the thermoelectric properties (Seebeck coefficient, electrical conductivity, electronic thermal conductivity) have been studied as a function of temperature and pressure up to 1200 K. The results show that Mg2Si is an n-type semiconductor with a band gap of 0.21 eV. The negative value of the Seebeck coefficient at all pressures indicates that the conduction is due to electrons. With the increase in pressure, the Seebeck coefficient decreases and electrical conductivity increases. It is also seen that, there is practically no effect of pressure on the electronic contribution of thermal conductivity. The paper describes the calculation of the lattice thermal conductivity and figure of merit of Mg2Si at zero pressure. The maximum value of figure of merit is attained 1.83×10−3 at 1000 K. The obtained results are in good agreement with the available experimental and theoretical results.
Thermoelectric based generators and refrigerators are solid-state devices in which heat energy is converted into electricity and vice versa. Thermoelectric generators have been used in power generation and remote power sources.[1] Thermoelectric generators are noiseless, reliable, and have no moving parts. Now, these generators are used in aerospace and military applications. Several thermoelectric materials based on tellurium, lead, antimony, and selenium has been investigated. These materials are more efficient because of high figure of merit, but are not safe to handle because of their toxic nature. These materials are less abundant and more expensive. Thus, it is a great challenge to search for new thermoelectric materials that are low cost, non toxic, and environment friendly. Semiconducting alkaline earth metal silicides have been of great interest due to their applications in thermoelectric devices.[2,3] Magnesium silicide and related alloys are promising material for thermoelectric devices because of their non toxic nature, thermal stability, low density, relative abundance, and low cost of production. Mg2Si material has been considered as a high performance thermoelectric material in the temperature range of 500 K–800 K.[4,5] Due to high specific strength and elastic modulus, Mg2Si can be used in the automobile and aerospace industry.[6,7] This material can also be used for optical fibers because it has good ohmic contact with Si.[8] Magnesium silicide is an n-type semiconductor having a band gap of 0.37 eV–0.78 eV.[9,10]
The parameter which evaluates the performance of thermoelectric materials is a dimensionless figure of merit ZT,[11] which is defined as


Efficient thermoelectric materials have high thermopower and low thermal conductivity. High performance thermoelectric materials have ZT equal to or greater than one. Baranek et al.[13,14] reported electronic, elastic, and phonon properties of this compound at zero pressure. Their studies have shown that the band gap is greater than the experimental value and all the electron calculations point out a mixed ionic–covalent nature of Mg–Si bonding. Tani and Kido[15] have reported the structural, elastic, and thermodynamic properties of this material at zero pressure. Several recent studies have reported the ZT value of Mg2Si and its solid solutions. Zaitsev et al.[16] achieved a maximum ZT of 1.1 within the temperature range of 300–870 K. Isoda et al.[17] achieved a maximum ZT, 1.2 at 620 K. Jung and Kim obtained ZT= 0.7 at 830 K with Bi-doped Mg2Si.[18] Kyung–Ho et al.[19] obtained maximum ZT= 0.24 at 773 K. Mao et al.[20] have experimentally reported that at 7.5 GPa, the cubic antiflourite structure changes into an anti-cotunnite structure. Yu et al.[21] reported the electronic, elastic, and thermal properties (entropy and specific heat) of Mg2Si at different pressures and temperatures. With the first-principles calculation, Yu et al.[22] reported the metallization of Mg2Si takes place at 8 GPa. They also found that the phase transformation from antiflourite to anti-cotunnite structure takes place at 8.38 GPa. Hao et al.[23] and Hao[24] experimentally revealed that a phase transition takes place at 7.5 GPa. Murtaza et al.[25] reported the effect of pressure on the electronic properties of this material. To the best of our knowledge, the thermoelectric properties such as the Seebeck coefficient, electrical and thermal conductivity, etc., of Mg2Si at different pressures and at high temperature has not yet been reported.
Therefore, the aim of this study is mainly devoted to predict the evolution of electronic and thermoelectric properties of Mg2Si under pressure to fill the lack of experimental data. The rest of the paper is divided into three sections. In Section 2, the computational methodology used in the present calculations is briefly explained. In Section 3, the results of electronic and thermoelectric properties of the material are discussed. Section 4 briefly summarizes the whole paper.
Mg2Si is the face-centered cubic (FCC) structure. Its corresponding space group is Fm-3m.[26] It belongs to the antifluorite structure family. There are three in-equivalent sites that can be specified in the irreducible unit cell, namely Si: a(0,0,0), Mg: a(1/4,1/4,1/4), and Mg: a(1/4,1/4,3/4). In these calculations, 2×1×1 super cell of Mg2Si which contains 24 atoms (16 Mg atoms and 8 Si atoms) has been constructed.
In this study, we adopted the same methodology which we have used to find the electronic and thermoelectric properties of Mg2C.[27] The electronic structure calculations were performed using density functional theory (DFT) based on the plane wave pseudo potential method as implemented in the Quantum Espresso package.[28] The generalized gradient approximation (GGA)[29] of Perdue–Burke–Ernzerhof (PBE) was used for the exchange–correlation functional. The cutoff for the kinetic energy was set to 40 Ry (1 Ry = 13.6056923 eV) for the plane-wave expansion of the electronic wave functions. The charge-density cutoff was kept at 400 Ry and the Marzari-Vanderbilt cold smearing size was fixed at 0.003 Ry. The Brillouin zone integration was performed using the Monkhorst–Pack scheme[30] with 4 × 8 × 8 meshes. The lattice constant of Mg2Si was optimized until the total energy converged to at least 10−6 Ry, and the forces between atoms became smaller than 10−4 Ry/bohr. For the density of state (DOS) calculation, we used the tetrahedron method with 16 × 16 × 16 denser k-point mesh.
We have interfaced the Quantum Espresso package with the BoltzTraP code[31] to calculate the thermoelectric properties. The BoltzTraP code is based on the Boltzmann theory and calculates various band structure dependent quantities such as electrical conductivity (σ) and electronic thermal conductivity (ke) within constant time approximation and rigid band approximation (RBA).[32,33] The BoltzTraP code can analytically represent these band energies with a smoothed Fourier interpolation and thereafter we can obtain the necessary derivatives such as electron velocities for transport properties. The electrical conductivity (σ) and seebeck coefficient (S) as a function of group velocity (vα) is expressed as
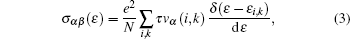

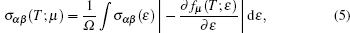

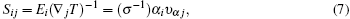

The electrical conductivity (σ) is expressed in terms of the ratio of σ/τ. To calculate the electrical conductivity of the system, we must determine the relaxation time. Hence, we adopted the strategy previously used by Ong et al.[34] and Zou et al.[35] We assumed that the relaxation time is direction independent, and treated the relaxation time as constant for certain specific temperature and carrier concentration. The reported experimental electrical resistivity (ρ) for Mg2Si is 7.14 × 10−2 Ω·cm[36] at 300 K, which combined with the calculated σ/τ yields τ = 0.176 × 10−14 s for this sample. By inserting these values into the standard electron phonon relaxation time equation (τ = CT−1n−1/3), the value of constant Cn−1/3 is determined as 5.28 × 10−13 s·K. Further, using this value, we calculate electrical conductivity σ.
In this study, we used the ShengBTE code[37] to calculate the lattice thermal conductivity, which is based on the phonon Boltzmann transport equation (pBTE). This code is based on the second-order (harmonic) and third-order (an harmonic) interatomic force constants (IFCs) combined with a full solution of the pBTE and can successfully predict the kl.[38,39] We calculated the second-order (harmonic) interatomic force constants (IFCs) from the Quantum Espresso package[28] and the third-order (an harmonic) interatomic force constants (IFCs) was obtained from ShengBTE.[37]
The lattice parameter and bulk modulus have been calculated by computing the total energy for different volumes and fitted to Murnaghan’s equation of state[40]

The calculated lattice constant, bulk modulus, and band gap at different pressures are summarized in Table
| Table 1. Calculated lattice constant, band gap, and bulk modulus at different pressures and comparison with available experimental and theoretical data. . |
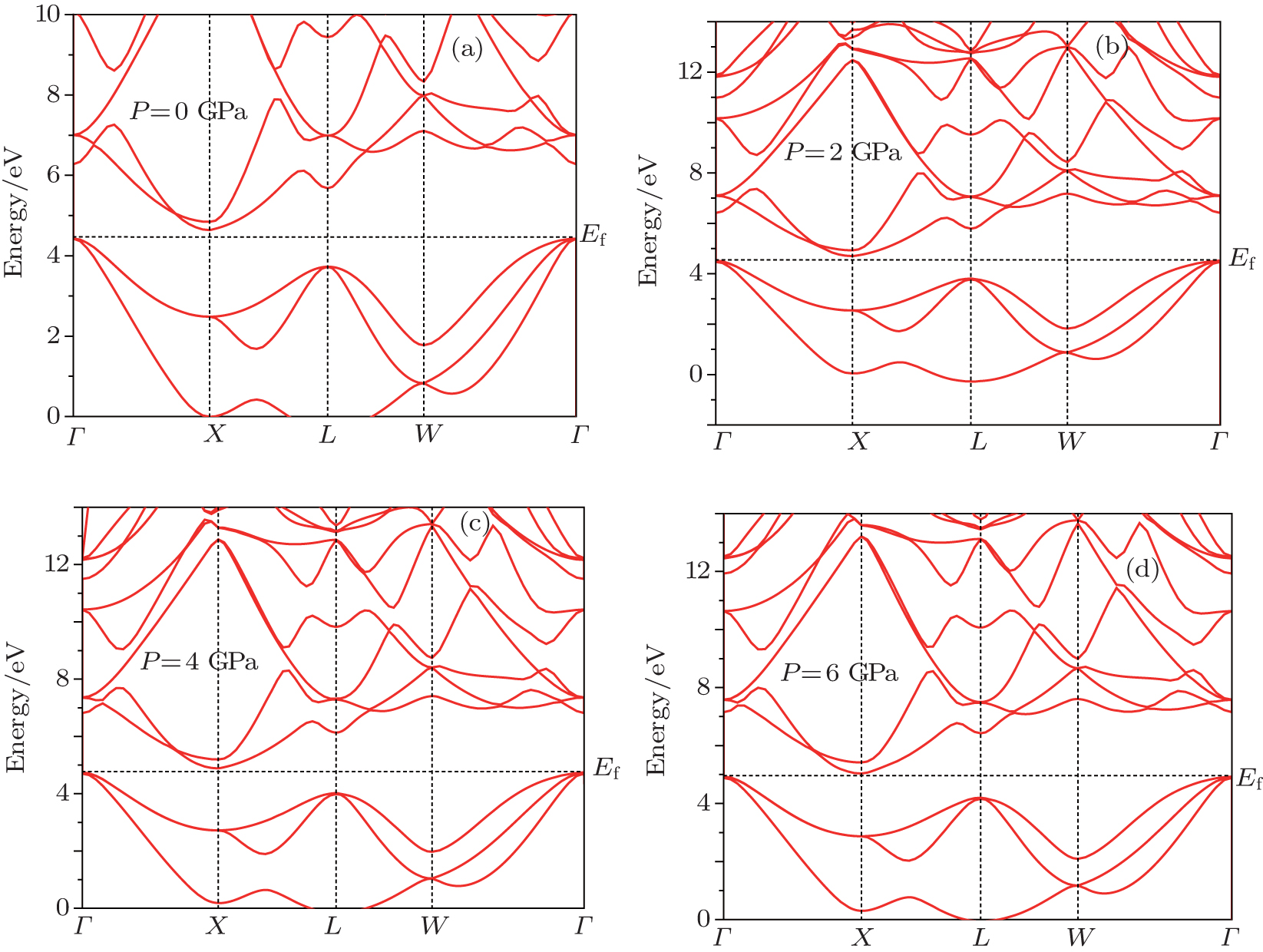 | Fig. 1. Electronic band structures of Mg2Si at different pressures. (a) P = 0 GPa, (b) P = 2 GPa, (c) P = 4 GPa, and (d) P = 6 GPa. |
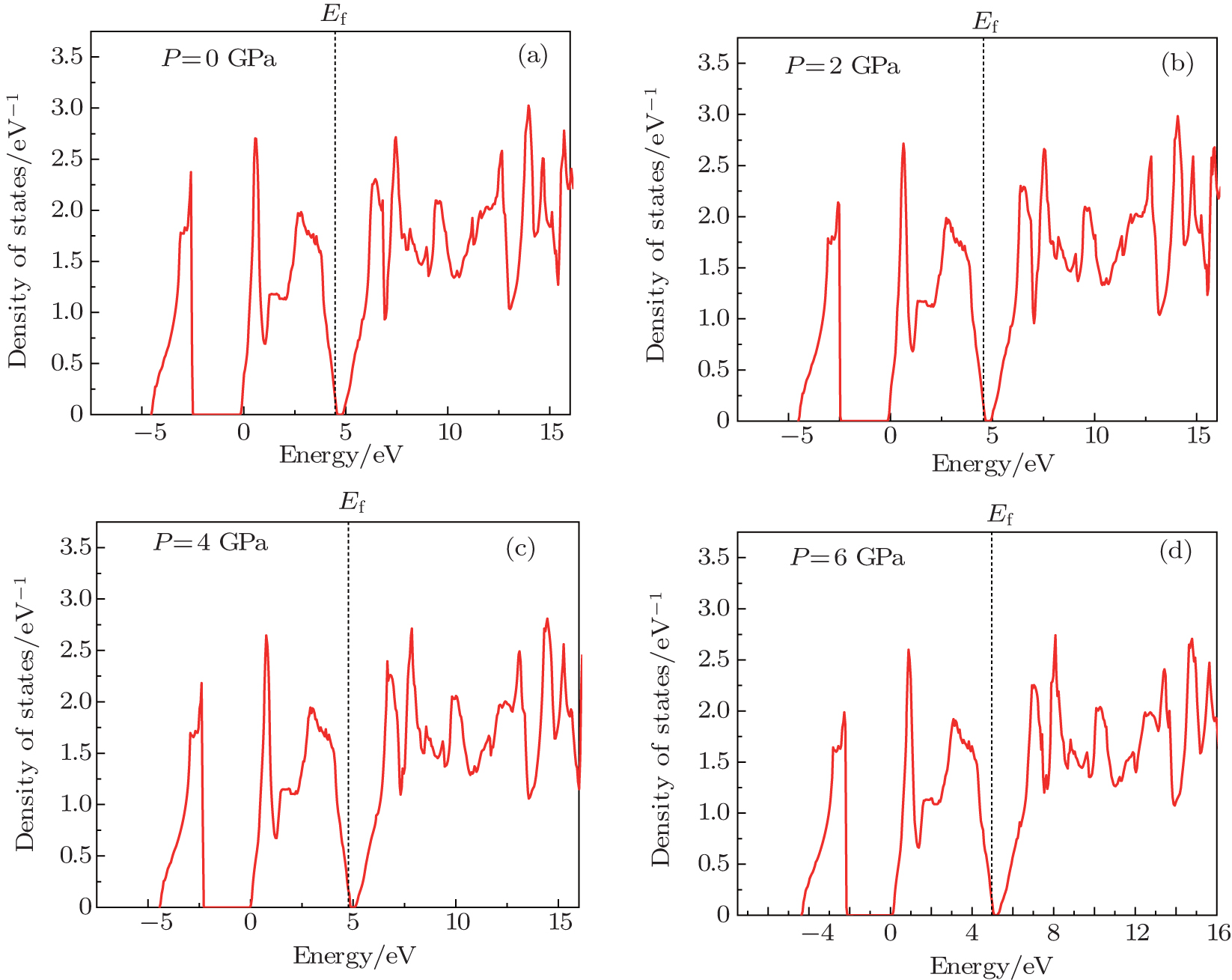 | Fig. 2. Density of states of Mg2Si at different pressures. (a) P = 0 GPa, (b) P = 2 GPa, (c) P = 4 GPa, and (d) P = 6 GPa. |
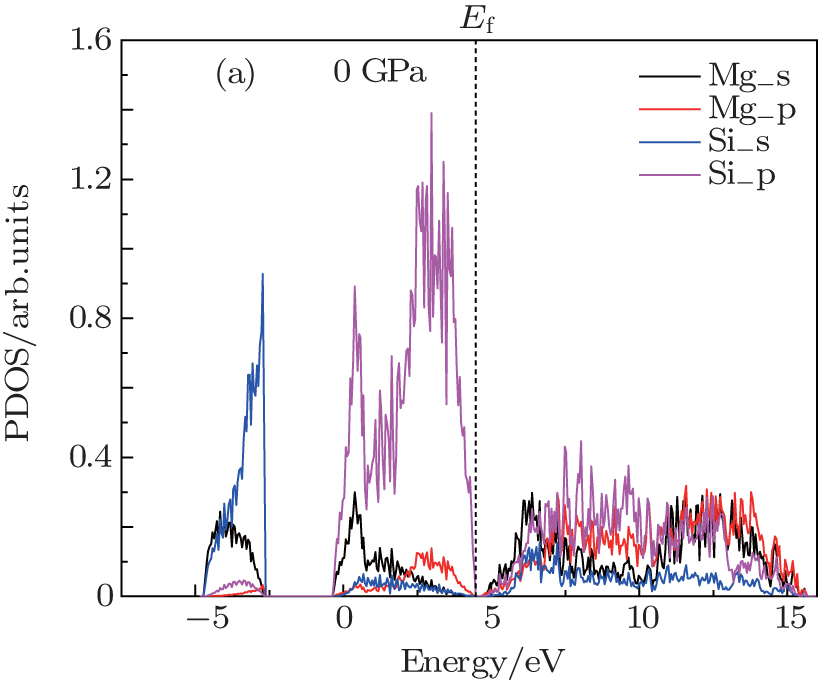 | Fig. 3. Projected density of state of Mg2Si at zero pressure. |
The variation of Seebeck coefficient (S), the electrical conductivity (σ/τ), the electronic thermal conductivity (ke/τ), and power factor (S2σ/τ) with respect to temperature are shown in Fig.
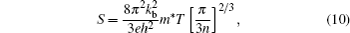
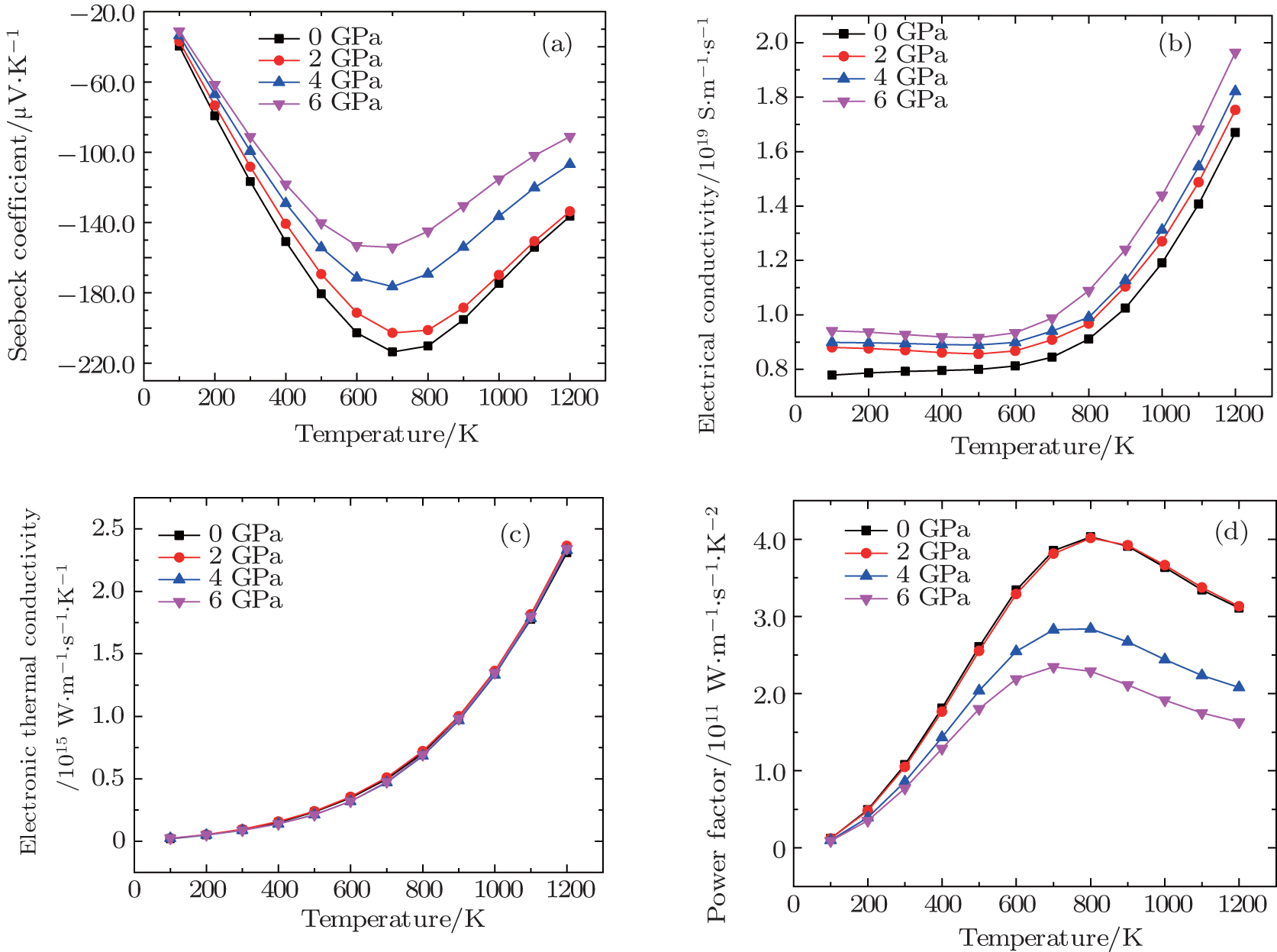 | Fig. 4. (a) Seebeck coefficient (S), (b) electrical conductivity (σ/τ), (c) electronic thermal conductivity (ke/τ), and (d) power factor (S2σ/τ) versus temperature at different pressures. |
Figure
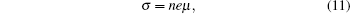
Figure

Figure
From the above discussion, we conclude that with an increase in pressure, the power factor of Mg2Si decreases. Power factor is an accumulated effect of Seebeck coefficient and electrical conductivity. With the increase in pressure, the band gap decreases which decreases the effective mass of electron. The Seebeck coefficient directly depends upon the effective mass, thus reduces the effective mass, decreases the Seebeck coefficient, and increases the electrical conductivity. Here, the Seebeck coefficient plays the main role in decreasing the power factor.
Figure
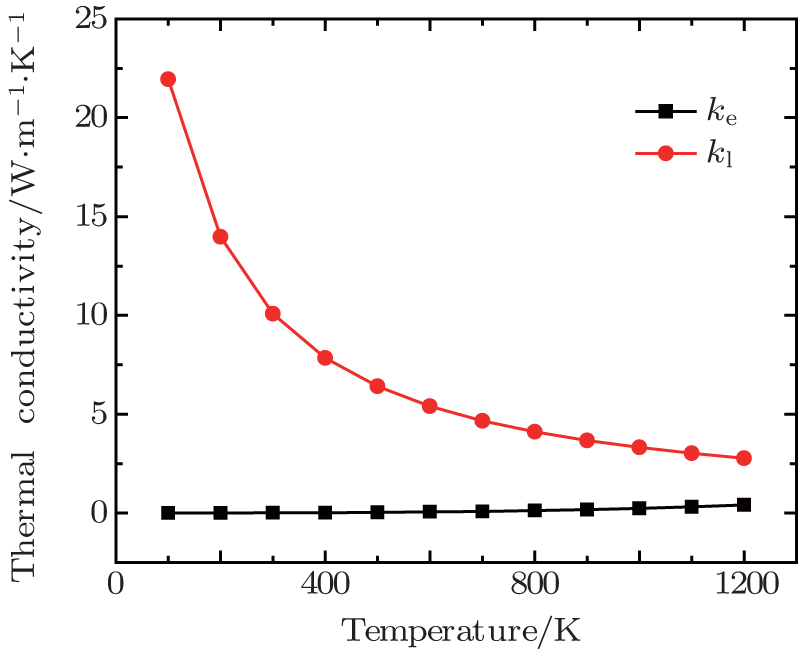 | Fig. 5. Electronic (ke) and lattice (kl) thermal conductivity versus temperature at zero pressure. |
 | Fig. 6. Variation of total thermal conductivity with temperature at zero pressure. |
Figure
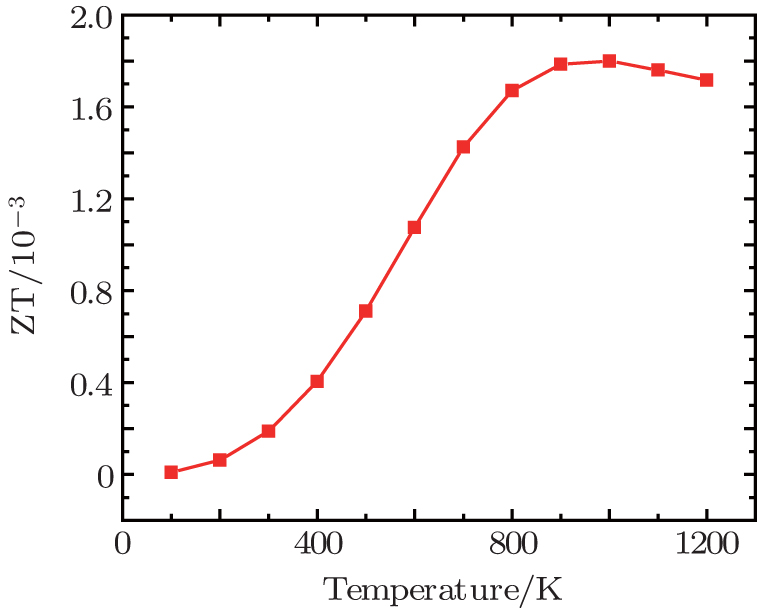 | Fig. 7. Figure of merit (ZT) versus temperature at zero pressure. |
We have used the density functional theory with GGA to investigate the effect of pressure on the electronic and thermoelectric properties of Mg2Si. The results show that the band gap decreases with pressure. Instead of electronic properties, overall thermoelectric properties decrease with pressure. The Seebeck coefficient and power factor decrease with pressure but electrical conductivity increases with pressure. The total thermal conductivity at normal pressure decreases with temperature and the maximum value of figure of merit is 0.0018 at 1000 K.
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 | |
| 22 | |
| 23 | |
| 24 | |
| 25 | |
| 26 | |
| 27 | |
| 28 | |
| 29 | |
| 30 | |
| 31 | |
| 32 | |
| 33 | |
| 34 | |
| 35 | |
| 36 | |
| 37 | |
| 38 | |
| 39 | |
| 40 | |
| 41 | |
| 42 | |
| 43 | |
| 44 | |
| 45 | |
| 46 | |
| 47 | |
| 48 | |
| 49 | |
| 50 | |
| 51 |