{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Lasing dynamics study by femtosecond time-resolved fluorescence non-collinear optical parametric amplification spectroscopy
[Dang Wei1, 2, Liao Qing3, Mao Peng-Cheng1, Fu Hong-Bing3, Weng Yu-Xiang1, †,  ]
]
]
|
|


† Corresponding author. E-mail:
Project supported by the National Natural Science Foundation of China (Grant Nos. 20925313 and 21503066), the Innovation Program of Chinese Academy of Sciences (Grant No. KJCX2-YW-W25), the Postdoctoral Project of Hebei University, China, and the Project of Science and Technology Bureau of Baoding City, China (Grant No. 15ZG029).
Femtosecond time-resolved fluorescence non-collinear optical parametric amplification spectroscopy (FNOPAS) is a versatile technique with advantages of high sensitivity, broad detection bandwidth, and intrinsic spectrum correction function. These advantages should benefit the study of coherent emission, such as measurement of lasing dynamics. In this letter, the FNOPAS was used to trace the lasing process in Rhodamine 6G (R6G) solution and organic semiconductor nano-wires. High-quality transient emission spectra and lasing dynamic traces were acquired, which demonstrates the applicability of FNOPAS in the study of lasing dynamics. Our work extends the application scope of the FNOPAS technique.
Lasing[1,2] and cooperative emissions (super-radiance[3] and superfluorescence[4,5]) are two types of coherent emissions. Lasing and cooperative emissions all feature a fast decay component in their radiative dynamics.[6–9] The evolution of emissive state, induced by energy transfer[10] or band gap renormalization,[11] is another important property with coherent emission. With respect to emission spectrum, the evolution of an emissive state would affect peak position and spectral shape. Time-resolved photoluminescence provides information on the radiative dynamics of an excited state directly for coherent emission. Among various time-resolved fluorescence spectroscopies, streak camera and fluorescence up-conversion technique have been extensively utilized in the study of coherent emission. The streak camera can provide an excellent spectrum coverage, but the poor time resolution is beyond one picosecond. Fluorescence up-conversion technique only provides the spectrum coverage of 10–20 nm in the collinear sum-frequency style,[12] while its time resolution is up to the limit of laser pulse duration. Compared to streak camera and fluorescence up-conversion technique, FNOPAS technique combines features of high time resolution and broad spectrum coverage together.
In 2005, Fita reported the first proof-of-principle experiment for FNOPAS.[13] This technique gates fluorescence photons and amplifies them in energy through a non-collinear optical parametric amplification process, which endows the FNOPAS with high sensitivity, broad detection bandwidth, and sub-picosecond time resolution. With the low detection limit, about 19 non-coherent fluorescence photons[14] and a single photon[15] for a coherent light source, have been reported in the condition of 150 fs gating width. With the broad detection bandwidth, the FNOPAS can afford a uniform gain bandwidth of 2500 cm−1 when using a 2-mm-thick β-BaB2O4 (BBO) crystal.[16] Moreover, an intrinsic spectrum correction function can be extracted from the parametric superfluorescence spectrum of FNOPAS.[16] By use of this intrinsic spectrum correction function, distortions of transient fluorescence spectrum are well corrected.
In this letter, the FNOPAS is employed to resolve the lasing process, one type of coherent emissions. First, the lasing from R6G dye solution in micro-cell was measured. The evolution of transient emission spectrum, laser build-up time, and feedback for lasing process were well demonstrated. The lasing dynamics of organic semiconductor nano-wires were also acquired by FNOPAS, including high-quality transient emission spectra and lasing dynamic trace. Our experimental results confirm the applicability of FNOPAS in the study of lasing dynamics.
The R6G ethanol solution (3 × 10−3 mol/L) fills a quartz micro-cell with an optical length of 1 mm. Two windows of the micro-cell are parallel to each other. Most stimulated emission photons transmit through the window of micro-cell, while a few of them are reflected by these two windows to undergo multiple gains. 1,4-chloride-2,5-di[4’-(methlthio)styryl-benzene (CDSB) nano-wire is prepared by the solution self-assembly method.[17] In the nano-wire, the CDSB molecules are arranged along the [100] direction to form a monoclinic crystal. The facet of the CDSB nano-wire displays a good rectangular shape (width: W = 1–2 μm; height: H = 0.5–0.8W), and uniformly distributes along the entire length (L) of several to several tens of micrometers. The detailed characterizations of CDSB nano-wire have been provided in Ref. [9]. The CDSB nano-wire and its two facets constitute a Fabry–Pérot microcavity, demonstrated by a set of resonant cavity modes.[9]
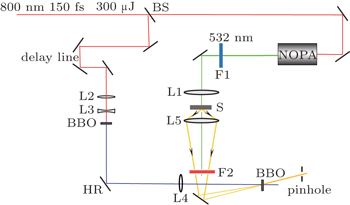
The experimental setup for the FNOPAS is schematically shown in Fig.
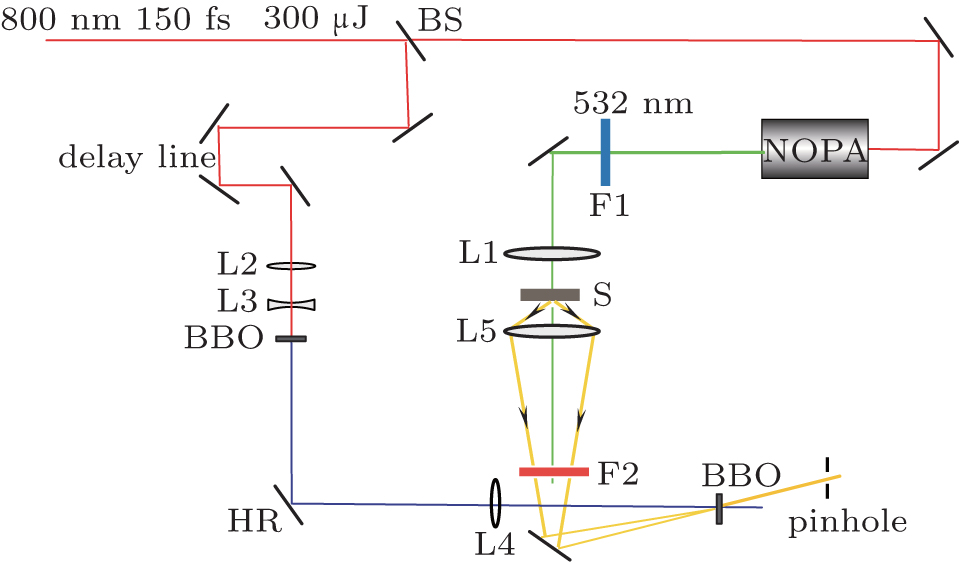 | Fig. 1. Setup of femtosecond time-resolved fluorescence non-collinear optical parametric amplification spectroscopy. BS: beam splitter; L1–L5: plano-convex lens; F1–F2: filter; S: sample; HR: high reflective mirror at 400 nm; NOPA: non-collinear optical parametric amplifier. |
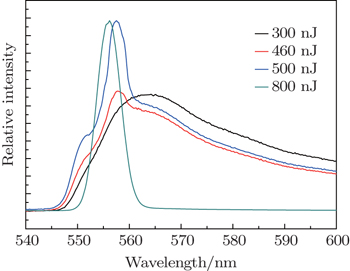
The time integrated emission spectrum of R6G solution displays a pump energy dependence, shown in Fig.
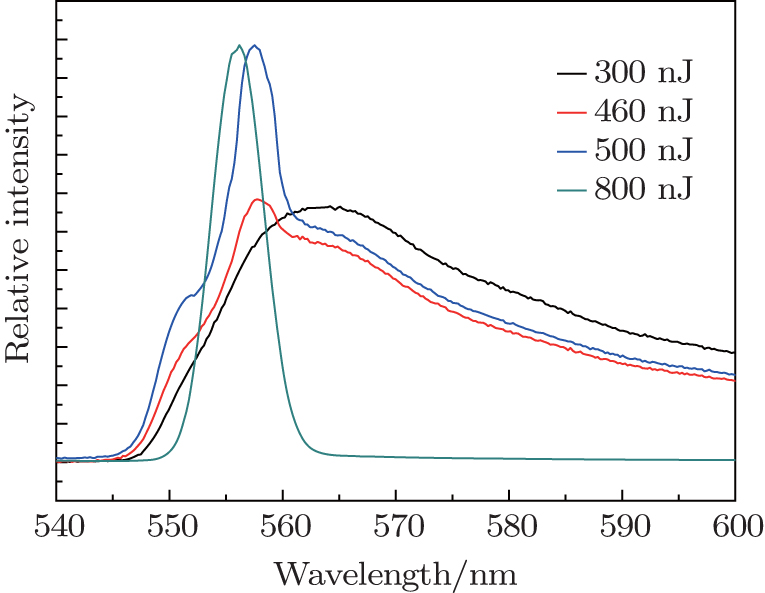 | Fig. 2. Time integrated emission spectra of R6G solution under four different pump energies: 300, 460, 500, 800 nJ per pulse. In the case of 800 nJ per pulse, the spectral intensity is scaled for comparison. |
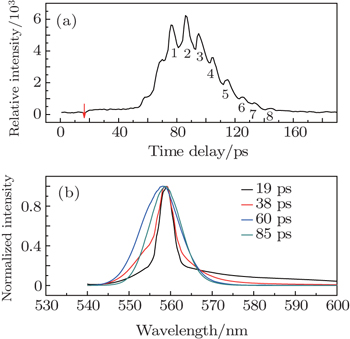
The lasing from R6G solution, under the pump energy of 800 nJ per pulse, was measured by the FNOPAS technique, shown in Fig.
Corresponding to the lasing dynamic trace (Fig.
More interestingly, there are eight peaks on the lasing dynamic trace (labeled with arabic number 1–8, at 76.5, 86.3, 95.1, 104.6, 114.6, 125.4, 134.6, 143.9 ps, see Fig. 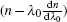
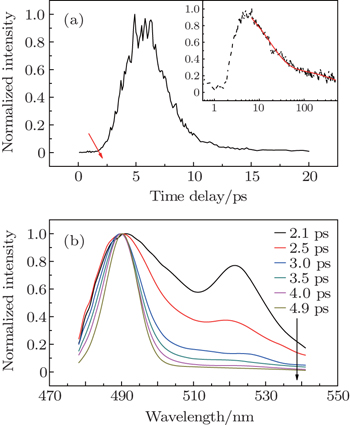
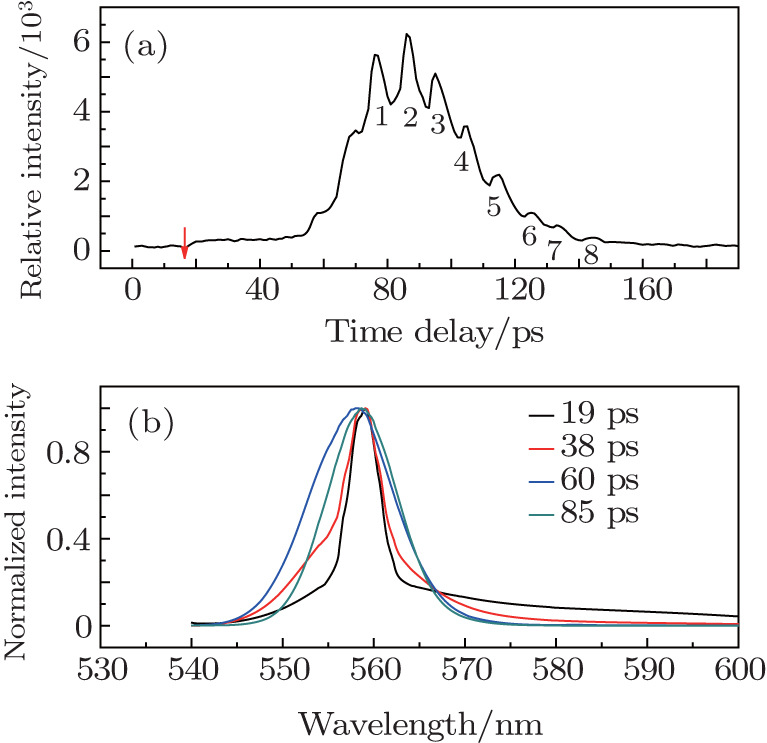 | Fig. 3. The lasing dynamic trace (556 nm) of R6G solution under the pump energy of 800 nJ per pulse (a) and its transient emission spectra (b). |
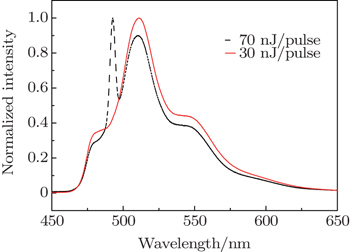
The above analyses have demonstrated the good performance of FNOPAS on the study of lasing from R6G solution. Now we extend the application of FNOPAS to the lasing dynamics of CDSB nano-wires. The spectral shape of CDSB nano-wires also depends on the pumping energy. Under weak pump energy (30 nJ per pulse), emission spectrum features the spontaneous emission (Fig.
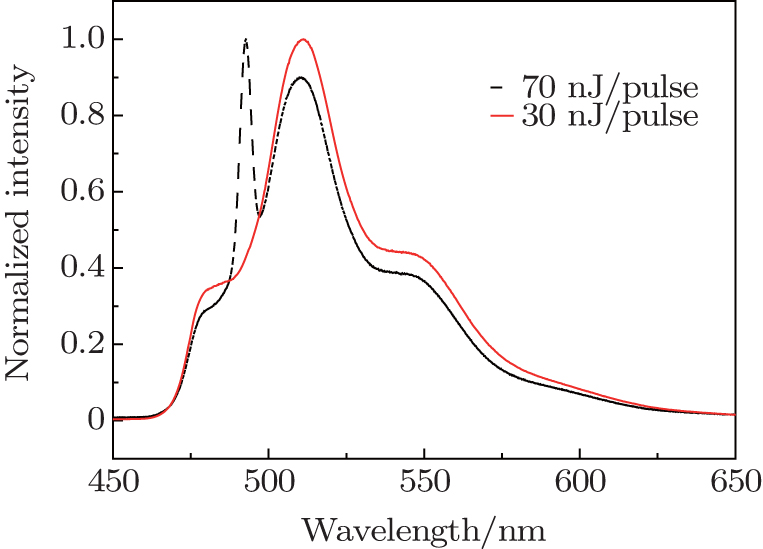 | Fig. 4. Normalized time integrated emission spectrum of CDSB nano-wires at 77 K. |
Under the pump energy of 100 nJ per pulse, transient emission spectra and lasing dynamic trace of CDSB nano-wires were measured (Fig.
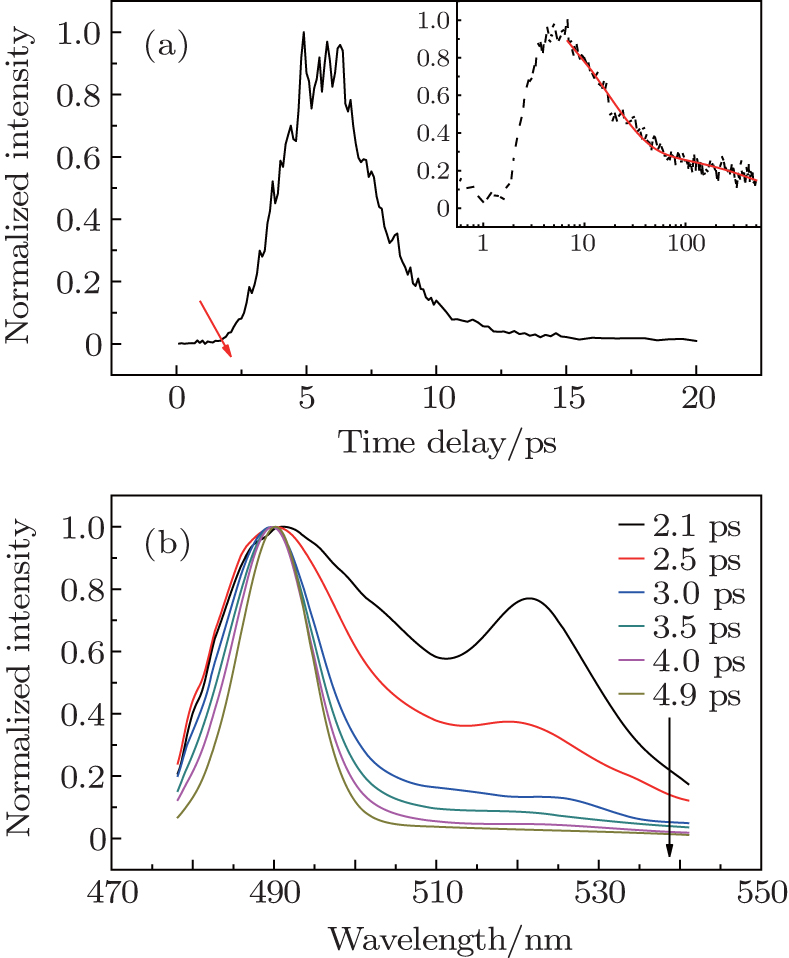 | Fig. 5. (a) The lasing dynamic trace (492 nm) of CDSB nano-wires. Inset: emission decay trace of CDSB nano-wires at 492 nm under the pump energy of 30 nJ per pulse (black dashed line) and its fitting (red solid line). (b) Transient emission spectra from CDSB nano-wires under the pump energy of 100 nJ per pulse (arrow direction represents the increase of time delay). |
Under the pump energy of 100 nJ per pulse, the lasing dynamic trace of CDSB nano-wires also exhibits some peaks (Fig.
The feasibility of FNOPAS in the study of lasing dynamics was examined in this letter. The evolution of transient emission spectrum, laser build-up time and feedback for lasing process were well demonstrated for R6G solution. Furthermore, lasing dynamics of CDSB nano-wires was also obtained by the FNOPAS. High-quality transient emission spectra and lasing dynamic trace provide access to the analyses of energy transformation in the organic semiconductor nano-wires. Our work suggests that the FNOPAS technique will be a powerful tool in the study of lasing dynamics.
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 | |
| 22 |