

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Graphene/polyaniline composite sponge of three-dimensional porous network structure as supercapacitor electrode
[Jiang Jiu-Xing, Zhang Xu-Zhi, Wang Zhen-Hua†,  , Xu Jian-Jun]
, Xu Jian-Jun]
, Xu Jian-Jun]
|
|


† Corresponding author. E-mail:
Project supported by the Natural Science Foundation from Harbin University of Science and Technology and Harbin Institute of Technology.
As a supercapacitor electrode, the graphene/polyaniline (PANI) composite sponge with a three-dimensional (3D) porous network structure is synthesized by a simple three-step method. The three steps include an in situ polymerization, freeze-drying and reduction by hydrazine vapor. The prepared sponge has a large specific surface area and porous network structure, so it is in favor of spreading the electrolyte ion and increasing the charge transfer efficiency of the system. The process of preparation is simple, easy to operate and low cost. The composite sponge shows better electrochemical performance than the pure individual graphene sponge while PANI cannot keep the shape of a sponge. Such a composite sponge exhibits specific capacitances of 487 F·g−1 at 2 mV/s compared to pristine PANI of 397 F·g−1.
As a single layer of carbon atoms, graphene which was discovered in 2004 by Novoselov et al.[1,2] exhibits extraordinary electrical, thermal and mechanical properties.[3] Graphene sponge which has a three-dimensional (3D) porous network structure has a large specific surface area and a fast electron transportation, so it can be used as a supercapacitor electrode. Because of the compressibility of sponge, it can also be used for other purposes, such as the compressible supercapacitor and sensor, etc. However, the pure individual graphene sponge has unsatisfactory capacitance.[4] Nowadays, graphene/polyaniline (PANI) has been successfully considered to be one of the most promising electrode materials due to its easy synthesis and relatively high conductivity and lower cost than many other conducting polymers.[5,6] However, swelling and shrinkage of PANI will occur in the process of doping/de-doping. Such a behavior may result in mechanical degradation of the electrode and fading of the electrochemical performance, so PANI reveals the disadvantage of a low cycle life.[7,8] The combination of PANI with various porous carbon materials will not only reinforce the stability of PANI, but also maximize the capacitance value.[6] Moreover, due to the large specific surface area, graphene has been proved to be an attractive substrate material when compounded with polymers and inorganic particles for lithium ion batteries, electrochemical capacitors or in other fields.[9–11] Graphene can create a local electrically conducting network to improve the specific capacitance and electrochemical cyclic stability of the electrode.[12,13] Therefore, the incorporation of graphene is highly favorable to synthesize nanoscale PANI. However, there is still a great challenge to the full use of the large surface area of graphene layers. The problem is that graphene may easily form irreversible agglomerates through van der Waals interactions.[14] By contrast, the oxygen-containing functional groups of GO facilitates stable dispersion in aqueous medium, in this case, it is easy to form a multilayer nanostructure of GO in solution.[12,15] Most researches show that the solution of graphene/PANI material was dried by a vacuum drying method, which only obtains powder or flake of the material.[6,9,14]
Herein, we report a simple three-step method to form the graphene/PANI composite sponge of 3D porous network structure. The composite sponge of graphene/PANI can macroscopically be seen as bulk, which is different from powder or flake, and microscopically, the composite sponge is of a 3D porous network structure. The 3D porous network structure is in favor of spreading the electrolyte ion and increasing the charge transfer efficiency of the system. Well dispersed deposition of PANI nanosheets on GO sheet could effectively increase the electrochemical utilization of PANI, and reduce the ion diffusion path during, that is, charge–discharge.[12] As a result, such a composite sponge exhibits better specific capacitance and cycling stability than pure graphene or PANI.
GO was synthesized from natural graphite by a modified Hummers method.[16] Firstly, graphite was turned into the expanded graphite by the method of acidification, drying and heating of a furnace tube. Then the expanded graphite was added into a 50-°C solution of concentrated H2SO4, concentrated H3PO4 and KMnO4, and the mixture was allowed to react for 6 h. After that, the reaction mixture was added into an ice water mixture and then stirred. In the final procedure, the mixture was treated with 30%-H2O2 until the color of the mixture became bright yellow to obtain a graphite oxide suspension. Finally the graphite oxide suspension was washed with 1:10 HCL solution and distilled water and centrifuged to obtain graphene oxide having a density of 8 mg/ml. The GO dispersion was centrifuged and the precipitate was directly dispersed in the solvent for the experimental step.
Exfoliation and dispersion of GO (200 mg) in 200 mL of distilled water were achieved by ultrasonication in an ultrasonic bath for 1 h. After that, aniline monomers (solvent: 1-M HCL) were added into the above suspension and sonicated for a moment. Afterwards, ammonium persulfate (APS, solvent: 1-M HCL) were rapidly added into the above mixture and stirred for 6 h in ice water mixture; the yellow–brown suspension gradually changed into a deep green color. Then the reactant was placed in a beaker and freeze-dried to obtain the composite sponge of GO/PANI. In this process, the molar ratio of aniline monomers to APS was 1:2 while the mass ratio of GO to aniline monomers was 1:0.4,1:1,1:3,1:5, etc. The composite sponge of GO/PANI was expressed as GOP, so the as-prepared products were named GOP1:0.4, GOP1:1, GOP1:3, GOP1:5. For comparison, the pure PANI and GO were also synthesized using a similar procedure to the above. The concentration of GO of all the composite GO/PANI material was 3 mg/ml.
The composite sponge of Graphene/PANI was obtained by reducing the prepared composite sponge of GO/PANI (from 2.1.2) in the steam environment of hydrazine at about 90 °C for 1 h. The composite sponge of graphene/PANI was expressed as RGP.
Scanning electron microscope (SEM) and infrared spectrum (FT-IR) equipment were used to characterize the textures, morphologies and chemical structures of GO, PANI, GOP and RGP. The SEM sample was prepared by using conductive adhesive pasting the sample onto a conductive copper plate. FT-IR spectra of films of GO, PANI, GOP, and RGP were recorded by a BRUKER TENSOR 27 FT-IR spectrometer with a smart OMNI sampler with potassium bromide crystal. X-ray photoelectron spectrum (XPS) measurement was performed using monochromator Al Kα irradiation. As suggested in the literature,[17] the electrochemical properties of the composite sponge of Graphene/PANI were characterized using a PARSTAT 4000 electrochemical working station with a three-electrode system in 2-M H2SO4 electrolyte at room temperature. The composite sponge of graphene/PANI served as the working electrode, while a platinum wire and an Ag/AgCl electrode were used as the counter electrode and the reference, respectively. The potential range for cyclic voltammetry (CV) measurements was from 0 V to 0.8 V at a scan rate of 2 mV·s−1.
The macroscopic appearance of all the samples except PANI (in Fig.
 | Fig. 1. Optical images of (a) sponge of GO, composite sponge of (b) GOP1:0.4, (c) GOP1:1, (d) GOP1:3, (e) GOP1:5, and (f) PANI power prepared by the process of freeze drying. |
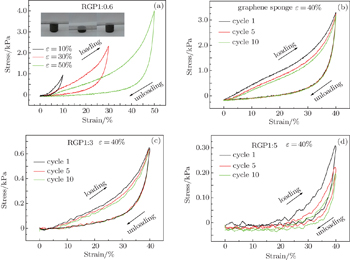
As shown in Fig.
 | Fig. 2. Compressive stress–strain curves of RGP1:0.6 (a), the stress–strain curves of graphene sponge (b), RGP1:3 (c), and RGP1:5 (d) at a maximum strain of 40% for 10 cycles. |
As shown in Figs.
 | Fig. 3. SEM images of ((a) and (b)) pure PANI at different magnifications. Both of them show the aggregations of nanofibers and nanoparticles mixture; SEM images of (c) GO, (d) GO/PANI. Each of them shows a 3D porous network structure; SEM images of ((e) and (f)) graphene/PANI at different magnifications. Each of them shows that graphene is evenly coated by PANI. |
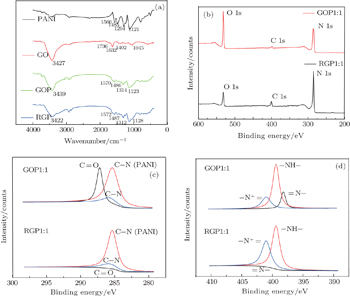
In the FT-IR spectrum of GO (Fig.
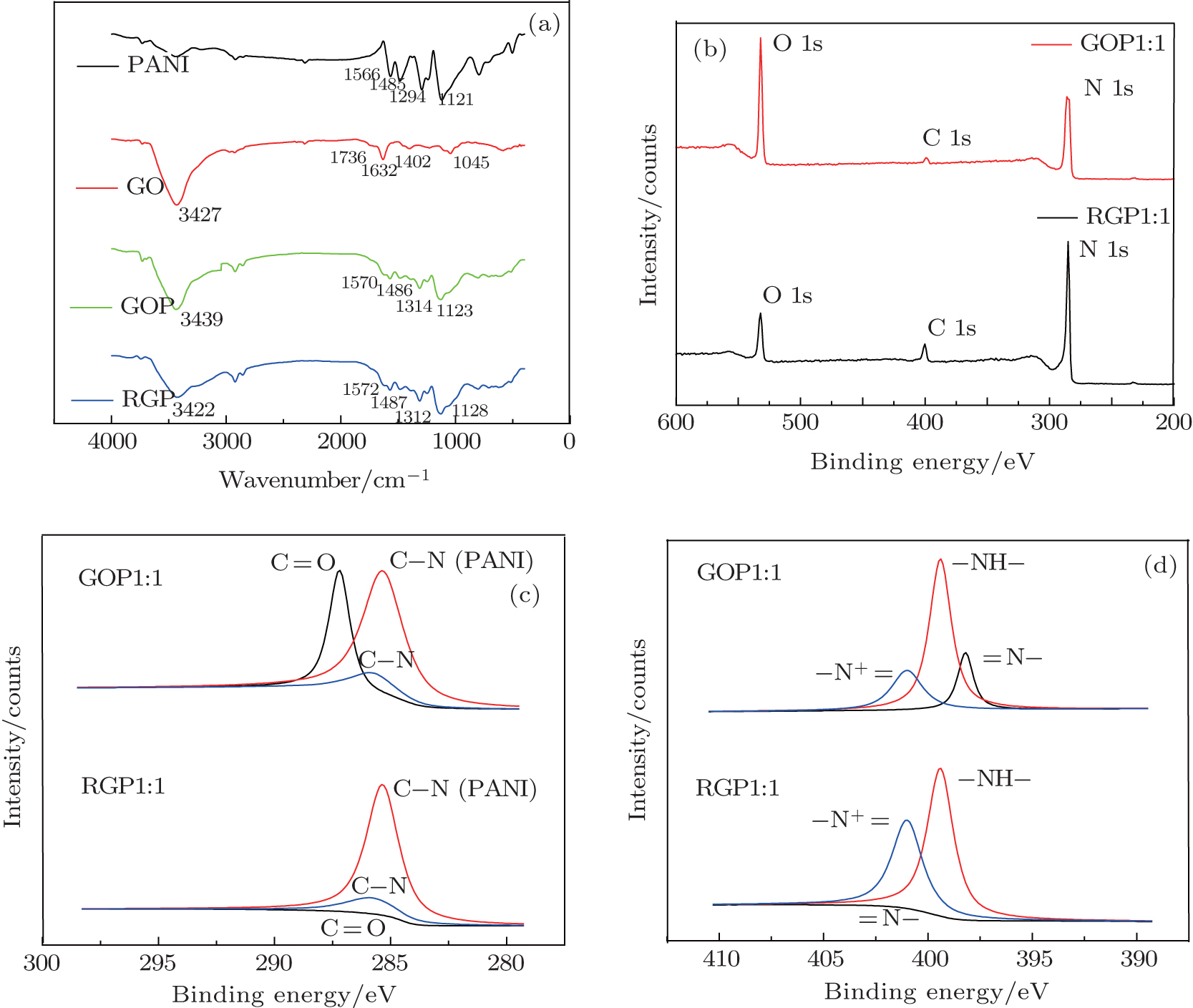 | Fig. 4. FT-IR (a) and XPS spectrum of GOP and RGP. (b) Elements in the spectrum. (c) C 1s core level spectrum of GOP and RGP. (d) N 1s core level spectrum of GOP and RGP. |
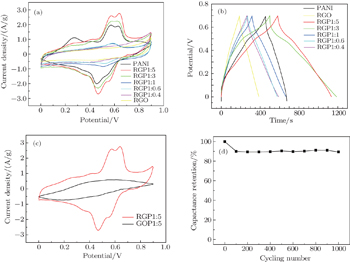
As is well known, the PANI deposited on carbon nanomaterials is found to be particularly advantageous to the supercapacitor because carbon material can provide a high surface area and excellent conductivity. This results in the deposition of nanostructured material with high specific capacitance.[29] The electrochemical performances of graphene/PANI materials as supercapacitor electrodes are investigated by using cyclic voltammetry (CV) and galvanostatic charge–discharge (GCD). Figure
 | Fig. 5. Electrochemical performances: (a) CV curves of PANI, RGP1:5, RGP1:3, RGP1:1, RGP1:0.6, RGP1:0.4, RGO, with the scan rate being 2 mV/s; (b) charge/discharge curves at a current density of 0.5 A/g; (c) CV curves of GOP1:5 and RGP1:5, with the scan rate is 2 mV/s; (d) cycling tests of 1000 charging and discharging cycles at 100 mV·s. |
Notably, as shown in Fig.
The 3D porous structure of the grapheme/polyaniline composite sponge is conducive to the diffusion of ion, and the surface of grapheme evenly coated with PANI exhibits short ion diffusion lengths, and good electrochemical utilization of PANI. Because of the compressibility of the sponge, it can also be used for other purposes, such as the compressible supercapacitor and sensors, etc. Of course, this also requires us to carry out further research. The electrochemical studies and SEM of composite sponge prove that graphene/PANI hybrid materials each possess a synergistic effect of PANI nanosheets and graphene. The composite sponge shows higher electrochemical capacitance, rate performance and cycling stability than each individual component. Therefore, the intriguing graphene/PANI composite sponge is quite a suitable and promising electrode material for supercapacitors.
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 | |
| 22 | |
| 23 | |
| 24 | |
| 25 | |
| 26 | |
| 27 | |
| 28 | |
| 29 | |
| 30 | |
| 31 | |
| 32 | |
| 33 | |
| 34 | |
| 35 |