Mahmood Q, Alay-e-Abbas S M, Mahmood I, Asif Mahmood, Noor N A. Investigations of mechanical, electronic, and magnetic properties of non-magnetic MgTe and ferro-magnetic Mg0.75TM0.25Te (TM = Fe, Co, Ni): An ab-initio calculation. Chinese Physics B, 2016, 25(4): 047101
Permissions
Investigations of mechanical, electronic, and magnetic properties of non-magnetic MgTe and ferro-magnetic Mg0.75TM0.25Te (TM = Fe, Co, Ni): An ab-initio calculation
Mahmood Q1, Alay-e-Abbas S M2, 3, Mahmood I4, Asif Mahmood5, Noor N A4, †,
Department of Physics, University of the Punjab, Quaid-e-Azam Campus, Lahore 54590, Pakistan
Department of Physics, Government College University Faisalabad, Allama Iqbal Road, Faisalabad 38000, Pakistan
Department of Physics, University of Sargodha, Sargodha 40100, Pakistan
Centre for High Energy Physics, University of the Punjab, Quaid-e-Azam Campus, Lahore 54590, Pakistan
College of Engineering, Chemical Engineering Department, King Saud University, Riyadh, Kingdom of Saudi Arabia
The mechanical, electronic and magnetic properties of non-magnetic MgTe and ferro-magnetic (FM) Mg0.75TM0.25Te (TM = Fe, Co, Ni) in the zinc-blende phase are studied by ab-initio calculations for the first time. We use the generalized gradient approximation functional for computing the structural stability, and mechanical properties, while the modified Becke and Johnson local (spin) density approximation (mBJLDA) is utilized for determining the electronic and magnetic properties. By comparing the energies of non-magnetic and FM calculations, we find that the compounds are stable in the FM phase, which is confirmed by their structural stabilities in terms of enthalpy of formation. Detailed descriptions of elastic properties of Mg0.75TM0.25Te alloys in the FM phase are also presented. For electronic properties, the spin-polarized electronic band structures and density of states are computed, showing that these compounds are direct bandgap materials with strong hybridizations of TM 3d states and Te p states. Further, the ferromagnetism is discussed in terms of the Zener free electron model, RKKY model and double exchange model. The charge density contours in the (110) plane are calculated to study bonding properties. The spin exchange splitting and crystal field splitting energies are also calculated. The distribution of electron spin density is employed in computing the magnetic moments appearing at the magnetic sites (Fe, Co, Ni), as well as at the non-magnetic sites (Mg, Te). It is found that the p–d hybridization causes not only magnetic moments on the magnetic sites but also induces negligibly small magnetic moments at the non-magnetic sites.
In recent years, spintronics (spin electronics) has proven itself to be the most rapidly advancing field in material science: its major goal is to explore spin-dependent phenomena and applications to new and versatile device functionalities.[1] The basic aim of governing the individual spins, particularly in solid state materials, could only be attained with the proper manipulation of magnetization and the spin currents. Nowdays, virtually all families of material are involved in spintronic research, however, the most potential candidates are the multilayer materials. In fact, the successful application of spin-dependent tunneling and scattering in such multilayer materials is reading heads of magnetic random access memories (MRAM) as well as high density hard discs.
Ferromagnetic semiconductors, in particular, appear to be more interesting due to the combination of ferromagnetic and semiconductor complementary functionalities. For example, powerful methods devised to govern the carrier concentration and spin polarization in semiconductor quantum paradigm could change the magnetization produced by the spin that is localized on the magnetic ions. Among ferromagnetic semiconductors, diluted magnetic semiconductors (DMSs) have received the greatest attention, as the local magnetic moments of doped magnetic impurities interact with the spin of charge carriers in the host semiconductors, thus making it possible to manipulate the spin degree of freedom of electrons. Therefore, ferromagnetic DMSs have been regarded as key and viable materials for spintronic applications.[2,3]
For more than two decades, the transition metal element doped IIb–VIb and IIa–VIb compounds have been widely studied.[4–9] These materials are categorized as magnetic and non-magnetic semiconductors. The magnetic semiconductors are equipped with properties different from those available in IIb–VIb and IIa–VIb binaries. These differences arise owing to the substitution of group-IIa and group-IIb cations for magnetic ions. The very special magnetic properties, owing to the addition of impurities, appear because of partially filled impurity 3d bands. The sp-bands of IIb–VIb and IIa–VIb binaries hybridize strongly with 3d bands of magnetic ion to give rise to technologically interesting electronic and magnetic properties. In earlier studies, the IIb–VIb and IIa–VIb based DMSs were investigated with Mn and Fe doping.[10–13] However, scientific interest has been aroused in recent years to study these materials by doping with other transition metals, like Co or Ni.[14–16]
Based on the above facts, one can propose the wide band gap IIa and IIb chalcogenides for studying the effects of transition metal dopants by using ab-initio calculations.[17–20] In the present work we pursue a study of the incorporation of transition metal (TM) elements Fe, Co and Ni dopant into wide band gap IIa–VI compounds MgTe, Mg0.75TM0.25Te (where TM = Fe, Co and Ni). To the best of our knowledge, no experimental as well as theoretical study has been conducted for these materials, which restricts the comparison of our results. However, this study will definitely provide a deep insight into the elastic, electronic and magnetic properties of these compounds to exploit them in the spintronics industry.
2. Computational details
All the results presented in this study were carried out within the framework of density functional theory (DFT), which has been successful in predicting the properties of materials with semiconducting as well as magnetic behaviors.[17–23] We have used the full potential linearized augmented plane wave plus the local orbital (FP-LAPW + Lo) method, which is implemented in the WIEN2K software.[24] We executed the calculations for equilibrium lattice parameters, bulk moduli and elastic constants by using the generalized gradient approximation (GGA) functional proposed by Perdew, Burke, and Ernzerhof (PBE).[25] On the other hand, the modified Becke–Johnson local density approximation functional (mBJLDA)[26] was used for electronic structure calculation and optical properties. The mBJLDA was used for electronic and optical properties due to the fact that it performs better in predicting the bandgap as compared with standard LDA[26,27] or GGA[23] exchange–correlation functional. The presence of 3d orbitals in the compounds under study requires going beyond standard GGA calculations. In this regard earlier studies[28] have shown that it is possible to improve results of GGA of compounds containing 3d elements by means of DFT +U calculations. In this method the value of U is chosen after performing tests for different values and selecting the ones which reproduce experimental results. However, it is also very well established that a particular value of U chosen for obtaining correct electronic properties fails to reproduce the structural properties,[28] thus making the choice of U dependent on the goal of study. Since in our work we performed a predictive study of mechanical, electronic and magnetic properties of the Mg0.75TM0.25Te (TM = Fe, Co, Ni) compounds, the implementation of GGA+U would require us to select the values of U for these calculations respectively. Moreover, the lack of experimental data on Mg0.75TM0.25Te (TM = Fe, Co, Ni) compounds does not guarantee that the GGA+U calculations can be effectively utilized. Since our calculated structural properties by using the GGA functional appear to be in good agreement with experimental data, an alternative functional is the mBJLDA which allows for the explorations of electronic and magnetic properties at the GGA calculated structural properties. In addition to its better performance for bandgaps, another important feature of the mBJLDA is its better treatment of the 3d orbitals, which have previously been shown by us[29] and other researchers[30] to produce the results consistent with experimental observations. Therefore, in the present study we use the GGA functional for computing elastic properties and structural parameters, while the electronic and magnetic properties are investigated by using the mBJLDA functional.
In order to achieve x = 0.25 dopant concentration of TM elements (Fe, Co, and Ni) in the zinc blende (ZB) MgTe semiconductor, we construct an eight-atom supercell of MgTe and then replace the Mg atom at the apex by the TM substituent. The considered concentration level leaves all the structures in cubic symmetry. In all calculations, the basis set convergence parameter RMT × Kmax (the product of the smallest of the atomic sphere radii RMT and plane wave cutoff parameter Kmax) is set to be 8. The maximum angular momentum quantum number lmax is set to be 10 inside the atomic sphere and Gmax is chosen to be 18.0 (Ryd)1/2. For accuracy of our calculations, a 2000 k−points sampling grid in the Brillouin-zone (BZ) integrations is utilized for both the binary and ternary compounds.
3. Results and discussion
3.1. Structural stability and mechanical properties
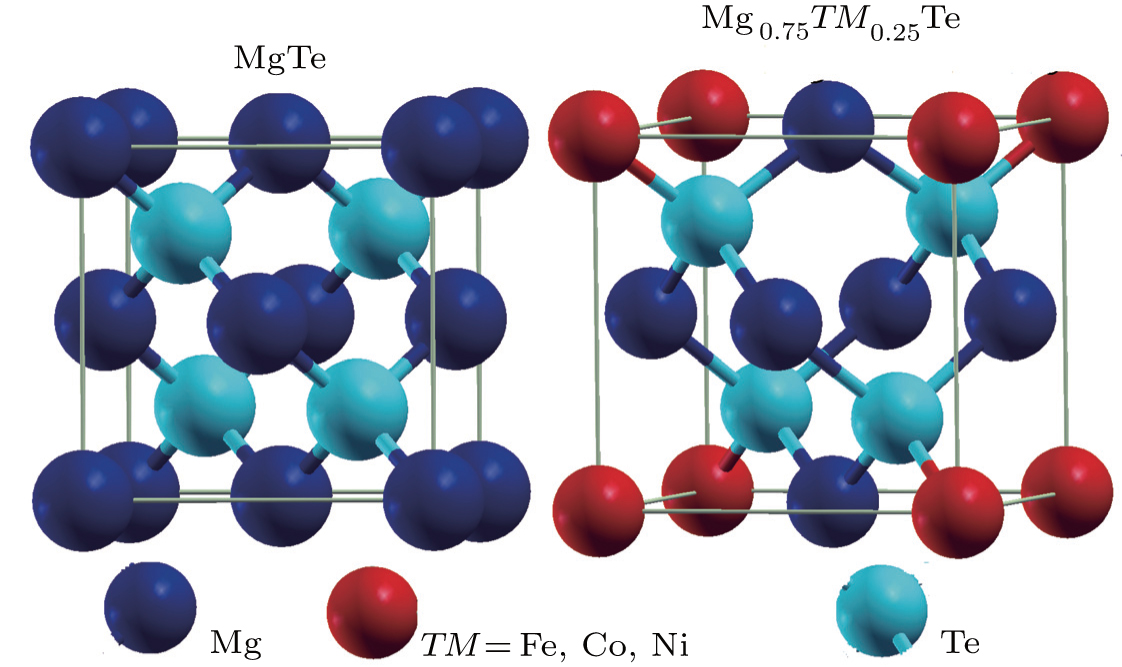
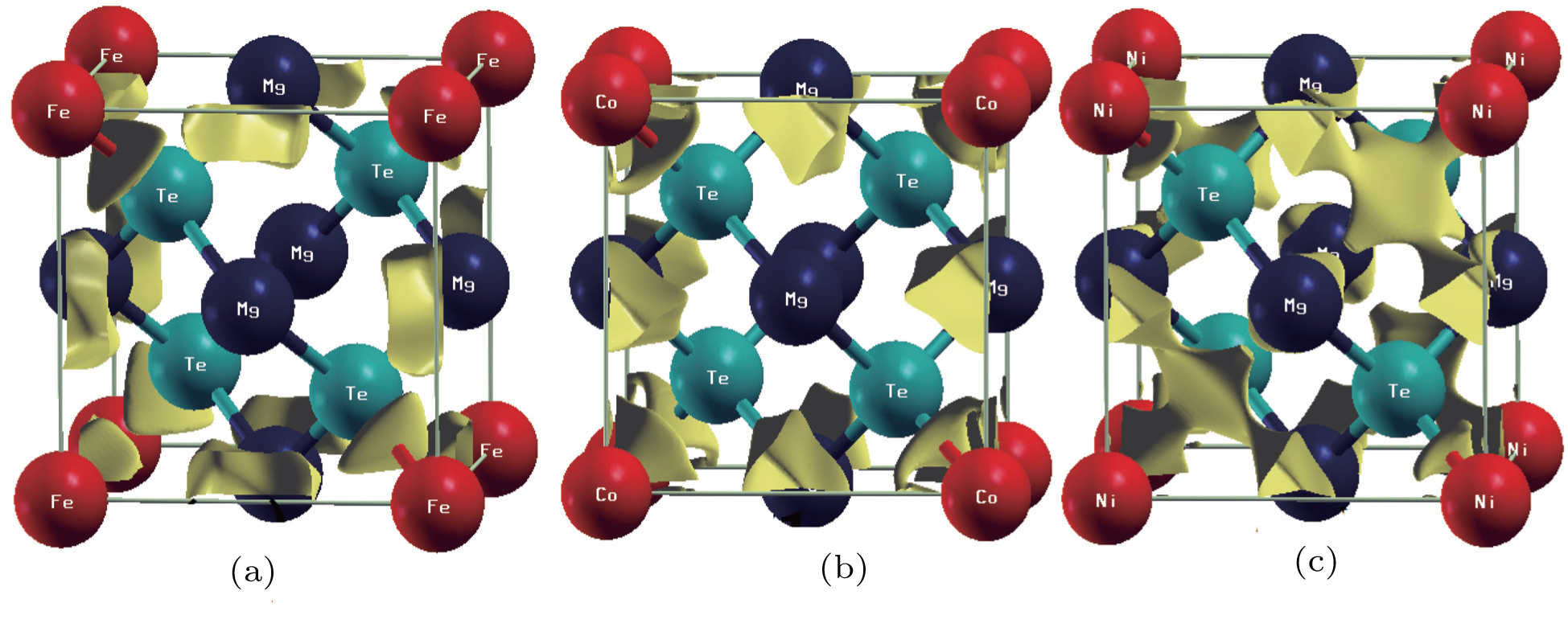
The conventional cells of zinc-blende (ZB) MgTe binary and Mg0.75TM0.25Te (TM = Fe, Co, Ni) ternary structures are given in Fig. 1. All the structures of alloys are fully relaxed in order to obtain ground state properties like optimized volume, Bulk modulus, lattice parameter and the enthalpy of formation as presented in Table 1. From Table 1 it is confirmed that our theoretically modeled results (lattice parameter and bulk modulus) have sound agreement with the experimental data.[31] However, the small overestimation and underestimation of the lattice parameter and bulk modulus (1.1% and 3.28%), respectively, of MgTe from experimental results are due to the use of the GGA functional.[23,26] The lattice parameters decrease with going from Fe doped MgTe to Ni doped MgTe which can be attributed to reducing the value of atomic radius of the dopant.
Fig. 1. Crystal structures of MgTe compound and its alloy Mg0.75TM0.25Te (TM = Fe, Co, Ni).
Table 1.
Table 1.
Table 1.
Calculated values of lattice constant a0 (in unit Å), bulk modulus B0 (in unit GPa), total energy difference ΔE1 (in unit eV) and ΔE2 (inunit eV), and enthalpy of formation ΔHf (in unit eV) for the MgTe compound and their alloys Mg1−xTMxTe (TM = Fe, Co, and Ni) at x = 0.25.
Calculated values of lattice constant a0 (in unit Å), bulk modulus B0 (in unit GPa), total energy difference ΔE1 (in unit eV) and ΔE2 (inunit eV), and enthalpy of formation ΔHf (in unit eV) for the MgTe compound and their alloys Mg1−xTMxTe (TM = Fe, Co, and Ni) at x = 0.25.
.
In order to investigate the preferred magnetic order in which the studied compounds are stable, we examine the ground state total energies of Mg0.75TM0.25Te (TM = Fe, Co, Ni) in antiferromagnetic (AFM), paramagnetic (PM) and ferromagnetic (FM) phases. The positive values of total energy differences ΔE1 = EPM − EFM and ΔE2 = EAFM − EFM show that the ternary compounds are stable in the FM phase (the energy difference values are also listed in Table 1). We also calculate the difference in the total energy between 32-atom supercells having FM and AFM magnetic orders. The calculated energy difference values (0.0079 eV, 0.0078 eV, and 0.0058 eV) for a supercell containing 32-atoms of alloys Mg0.25Fe0.75Te, Mg0.25Co0.75Te, and Mg0.25Ni0.75Te show that the FM ordering between magnetic ions becomes weaker outside the tetrahedral environment of nearest neighbors. This confirms the short range FM ordering in these compounds, however, FM remains stable as compared with AFM. To further confirm the stabilities of these compounds in the ferromagnetic phase we calculate the enthalpies of formation for Mg0.75TM0.25Te (TM = Fe, Co, Ni) in the FM phase by using the relation
where Etotal (MgTe : TM), ETM = Fe,Co,NiEMg, and ETe are the calculated total energies of cubic Mg0.75TM0.25Te (TM = Fe, Co, Ni), ground state energies per atom TM = Fe, Co, Ni (space group #217, I-43M), hexagonal Mg (space group #194, P63/mmc) and trigonal tellurium (space group #152, P3121), respectively. The calculating lower value of enthalpy of formation for MgTe and Mg0.75TM0.25Te (shown in Table 1) represents the energy released during compounds formation, which makes them stable in the FM phase. The reducing value of enthalpy of formation for Fe as compared with for Ni dopant indicates that Mg0.75Fe0.25Te is stabler than Mg0.75Ni0.25Te.
To explore the mechanical and dynamical behaviors of the compounds, the knowledge of elastic constants is of supreme importance. Moreover, elastic constants allow one to predict how a material would behave under the applied stress.[32] We perform ab-initio calculations to compute elastic modulii Ci j by calculating stress tensor components for small strain. These constants play an important role in providing valuable information about the stability and stiffness of material. The method used to evaluate these constants was developed by Wong et al.[22] which has already been successfully used in some previous studies.[33–35] Since each of the investigated materials is of cubic symmetry, it is necessary to calculate only three independent elastic parameters C11, C12, and C44 that can completely describe their mechanical properties. Hence, three equations are needed to calculate these constants.[36] The generalized Hooke’s law provides a connection between stresses (s) and strains (ɛ) to obtain elastic moduli. The bulk modulus, shear modulus, and Young modulus are given by
where
On the other hand the Kaleinmen parameter, Poisson ratio, and anisotropy constant are calculated from the following relations:
The Born stability criterion[37] determines the mechanical stability of a cubic crystal. The values of bulk modulus (B) estimated using the relation Eq. (2) and by energy minimization have nearly the same values as those given in Table 1, which confirms the reliability of calculated elastic properties of the compounds. The Kleinman parameter (ζ) depicts the dominant character of either bond bending or bond stretching in cubic material. A value close to 0 indicates bond bending, while a value approaching to 1 indicates bond stretching. From the data presented in Table 2, it is easy to see that all the binary and ternary compounds show bond stretching, which is increased with the introduction of dopant from Fe to Ni.[38]
Table 2.
Table 2.
Table 2.
Calculated values of elastic constants Ci j (in unit GPa), Kleinman parameters (ζ), shear modulus G (in unit GPa), Young’s modulus E (in unit GPa), Poisson’s ratio, Lames constants, anisotropy factor, shear wave modulus, and B/G ratio at equilibrium for the parent compound and their alloys Mg1−xTMxTe (M = Fe, Co, and Ni) at x = 0, 0.25.
Calculated values of elastic constants Ci j (in unit GPa), Kleinman parameters (ζ), shear modulus G (in unit GPa), Young’s modulus E (in unit GPa), Poisson’s ratio, Lames constants, anisotropy factor, shear wave modulus, and B/G ratio at equilibrium for the parent compound and their alloys Mg1−xTMxTe (M = Fe, Co, and Ni) at x = 0, 0.25.
.
In the field of engineering science, the knowledge of the elastic anisotropy is of the great importance as it provides information about the possibility of micro cracks to be introduced in a material.[39] In this context, we evaluate the anisotropy factor A by using the calculated elastic constants. For an isotropic material it is equal to 1, while any deviation (lower or higher) from 1 indicates anisotropy.[40] Referring to Table 2, all the compounds being investigated are anisotropic materials, as A undergoes deviation from unity.
For the evaluation of the shear modulus of the compound, the Voigt–Reuss–Hill[41] approximation is employed since the material under investigation is generally synthesized into polycrystal. According to this approximation, the calculated values can be driven from the arithmetic mean of the two well-known bonds for mono-crystals as suggested by Voigt,[42] and Reuss and Angew.[43] In this regard, the above mentioned shear modulii for cubic structures are worked out from the relations given in Ref. [40]. Moreover, Young’s modulus (E) and Poisson’s ratio (ν) are also calculated. Both of these factors are linked with the bulk modulus (B) and Shear modulus (G).[43,44] The estimated elastic moduli for the binary MgTe and its alloys Mg0.75TM0.25Te (TM = Fe, Co, Ni) are listed in Table 2. Higher value of E represents stiffer material as explained in the section about structural properties. Since the value of E increases with going from Fe to Ni, the alloy Mg0.75Ni0.25Te appears to be stiffer than Mg0.75Fe0.25Te and Mg0.75Co0.25Te. Among the calculated elastic parameters, ν is the most capable of explaining the nature of the bonding forces at work in a material. The upper and lower limits defined for ν are 0.5 and 0.25, respectively.[45] Since the value of ν lies within 0.25–0.5 for Mg0.75Fe0.25Te, Mg0.75Co0.25Te, and Mg0.75Ni0.25Te, so the central inter-atom forces are set in these alloys. The covalent and ionic materials can also be distinguished with ν, which is typically 0.1 and 0.50 for covalent and ionic materials, respectively. On the basis of estimated values of ν, MgTe and ternary counterparts are partially ionic. Furthermore, the critical value 0.26 of Poisson’s ratio separates a brittle material from a ductile one.[46] It is obvious from our calculated ν values that MgTe is brittle, while Mg0.75Fe0.25Te, Mg0.75Co0.25Te, and Mg0.75Ni0.25Te are ductile.
The ductile and brittle nature of a material can also be characterized by Pugh‘s ratio (B/G) whose critical value is 1.75. Referring to Table 2, the B/G value is 1.77 for Mg0.75Fe0.25Te, 1.78 for Mg0.75Co0.25Te, and 1.84 for Mg0.75Ni0.25Te, classifying these compounds as ductile. From Young‘s modulus and Poisson’s ratio, other exciting parameters named Lame‘s constants can also be calculated by using the relations given in Ref. [47]. The first and second Lame constants are λ and μ and their values are given in Table 2. For an isotropic system, it is easy to show λ = C12 and μ = C44. Since the alloys Mg0.75Fe0.25Te, Mg0.75Co0.25Te, and Mg0.75Ni0.25Te are strongly anisotropic, our results totally deviate from the above stated relations as also evidenced from the previous studies[38] (see Table 2).
The knowledge of Debye temperature is of great importance as it has a close relationship with various physical properties such as specific heat, elastic constants, and melting temperature. From the calculations of Young’s modulus, Bulk modulus, and shear modulus, the Debye temperature is evaluated.[48] In a polycrystalline material, the average sound velocity is approximately given by Ref. [49], and ml and ms denote the longitudinal and transverse sound velocities. The values of G and B from Navier’s equation[50] help to obtain the values of ml and ms. The sound velocities dependent on the elastic moduli (B and G) of a material decrease with the increase of TM doping amount in MgTe. The increase of average sound velocity further reduces the longitudinal and transverse velocities. Moreover, the average sound velocity affects the Debye temperature. For MgTe, Mg0.75Fe0.25Te, Mg0.75Co0.25Te, and Mg0.75Ni0.25Te compounds, the Debye temperature is found to increase 10.6 K, 11.78 K, 11.83 K, and 12.6 K, respectively. Owing to the unavailability of experimental data for elastic constants, our predicted elastic constants will draw the attention of experimentalists to explore these materials for spintronic applications.
3.2. Electronic properties
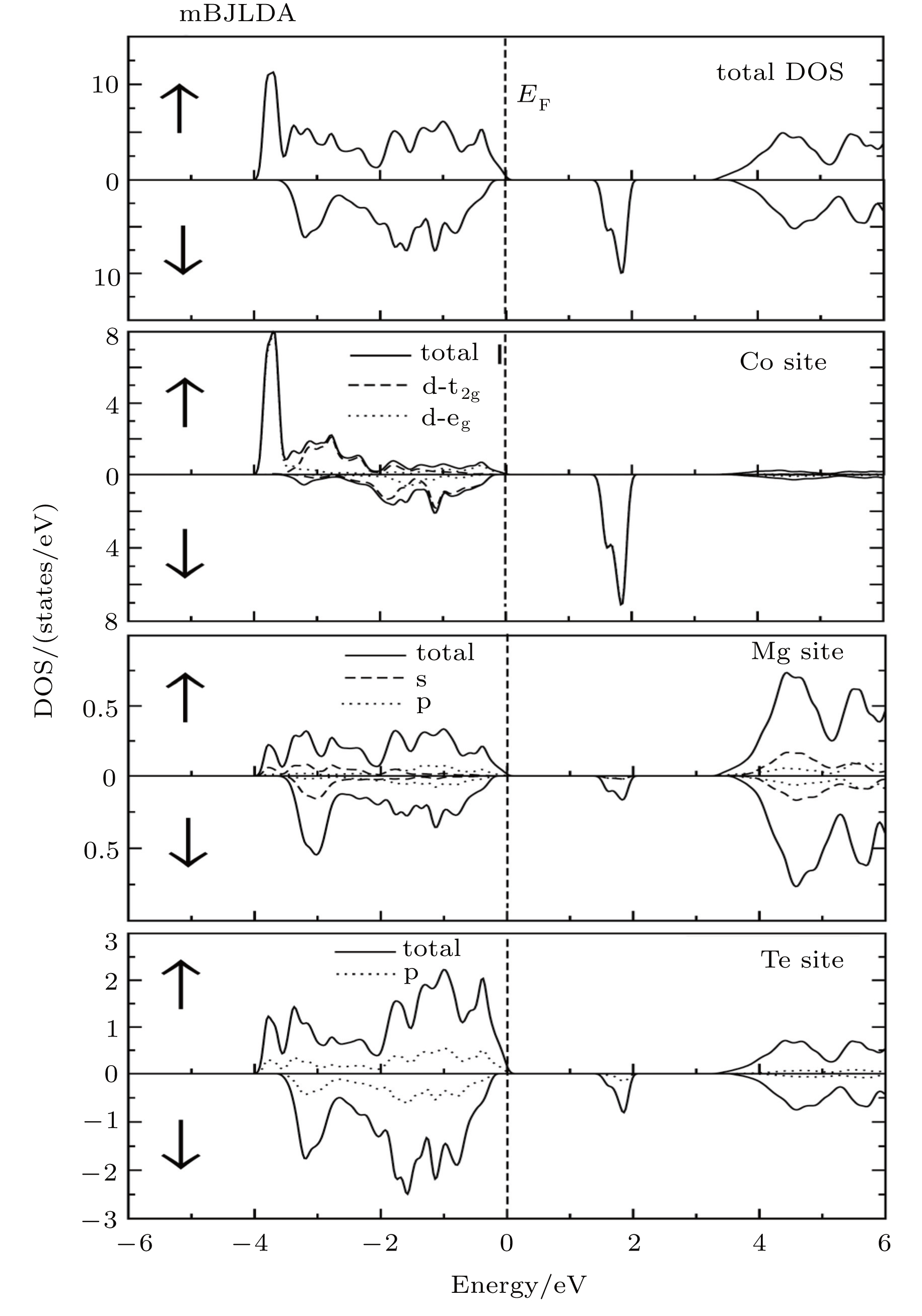
The spin-polarized electronic band structures, total (TDOS) and partial density of states (PDOS) and valence electron charge densities are computed for investigating the electronic properties and bonding natures of Mg0.75TM0.25Te (TM = Fe, Co, Ni) compounds. Owing to the fact that GGA calculations yield underestimated band gaps, we utilize mBJLDA for improving the band gap. The band structures and TDOS along with PDOS are presented in Figs. 2–5, respectively.
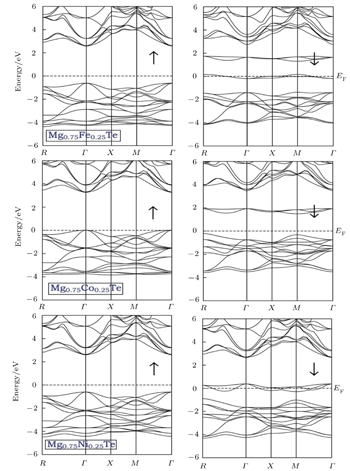
Fig. 2. Calculated spin polarized ferromagnetic band structures for Mg0.75TM0.25Te (TM = Fe, Co, Ni) with mBJ potential. The left panel is for majority spin (↑) and the right panel for minority spin (↓).
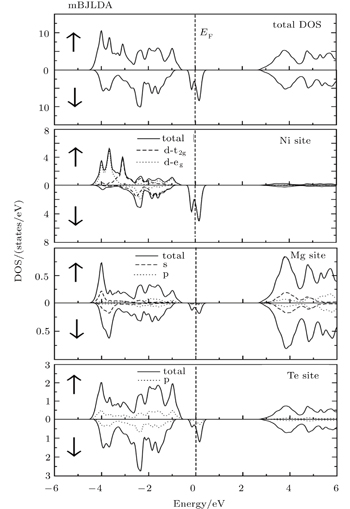
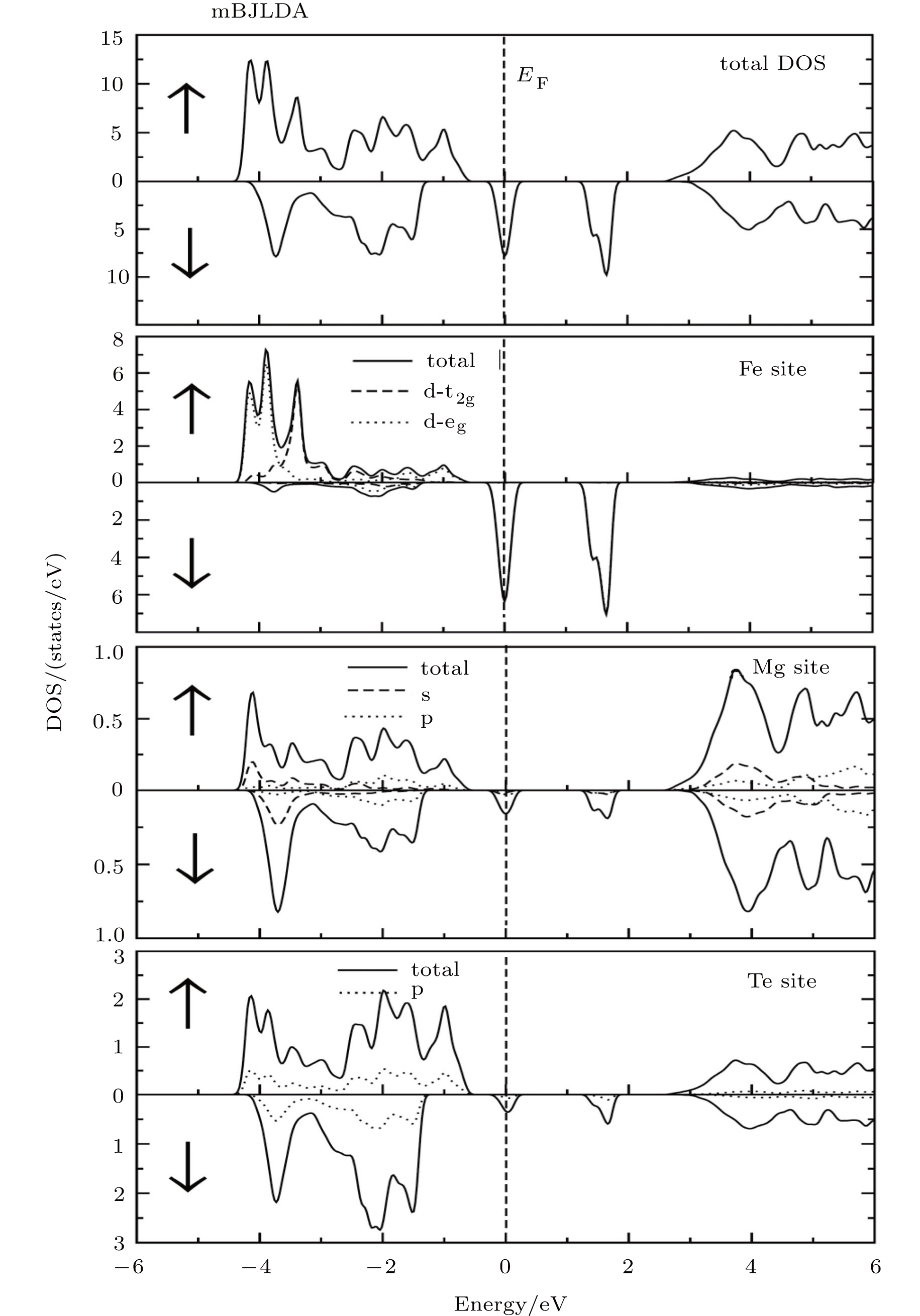
Fig. 3. Total densities of state (TDOSs) along with partial densities of state (PDOSs) of Fe site, Mg site and Te site for Mg0.75Fe0.25Te, obtained by using the mBJ potential for majority spin (↑) and minority spin (↓).
Fig. 4. Total densities of state (TDOSs) along with partial densities of state (PDOSs) of Co site, Mg site and Te site for Mg0.75Co0.25Te, obtained by using mBJ potential for majority spin (↑) and minority spin (↓).
Fig. 5. Total densities of state (TDOSs) along with partial densities of state (PDOSs) of Ni site, Mg site and Te site for Mg0.75Ni0.25Te, obtained by using mBJ potential for majority spin (↑) and minority spin (↓).
All the compounds demonstrate a direct band gap nature in the spin-up case, since the minima of CB and maxima of VB are both located at the Γ point. The band gap values for Mg0.75Fe0.25Te, Mg0.75Co0.25Te, and Mg0.75Ni0.25Te are 3.30 eV, 3.20 eV, and 3.10 eV, respectively. On the contrary, the spin-down band structure shows distinct features for TM-doped MgTe, which can be ascribed to differences in occupied 3d state among Fe, Co, and Ni atoms.
From the total density of states along with the partial density of states as shown in Figs. 3–5, it is evident that Te 5p states predominantly contribute to the upper and lower part of VB in both spin-up and spin-down cases, while the TM 3d states contribute more to the lower portion VB in an energy range from −2 eV to −4 eV. Like previous studies,[17] the s orbitals of Te, not shown in the partial density of state %PDOS plots, in the spin-down case are positioned slightly higher than that in the spin-up case. Moreover, the s-orbitals of Mg dominate the CB minima along with the contribution coming from the TM 4s states. In all the studied compounds, the VB has TM 3d-t2g states located slightly nearer to the Fermi level than TM 3d-eg states for the spin-up case, while these states show different behaviors for the spin-down channel. For the case of Mg0.75Fe0.25Te, both 3d-eg and 3d-t2g states are located above the valence band maxima in the spin-down band structure. On the other hand, the 3d-t2g states of Mg0.75Co0.25Te and Mg0.75Ni0.25Te are located inside the VB in the spin-down channel, while the 3d-eg states are located above and at the Fermi level.
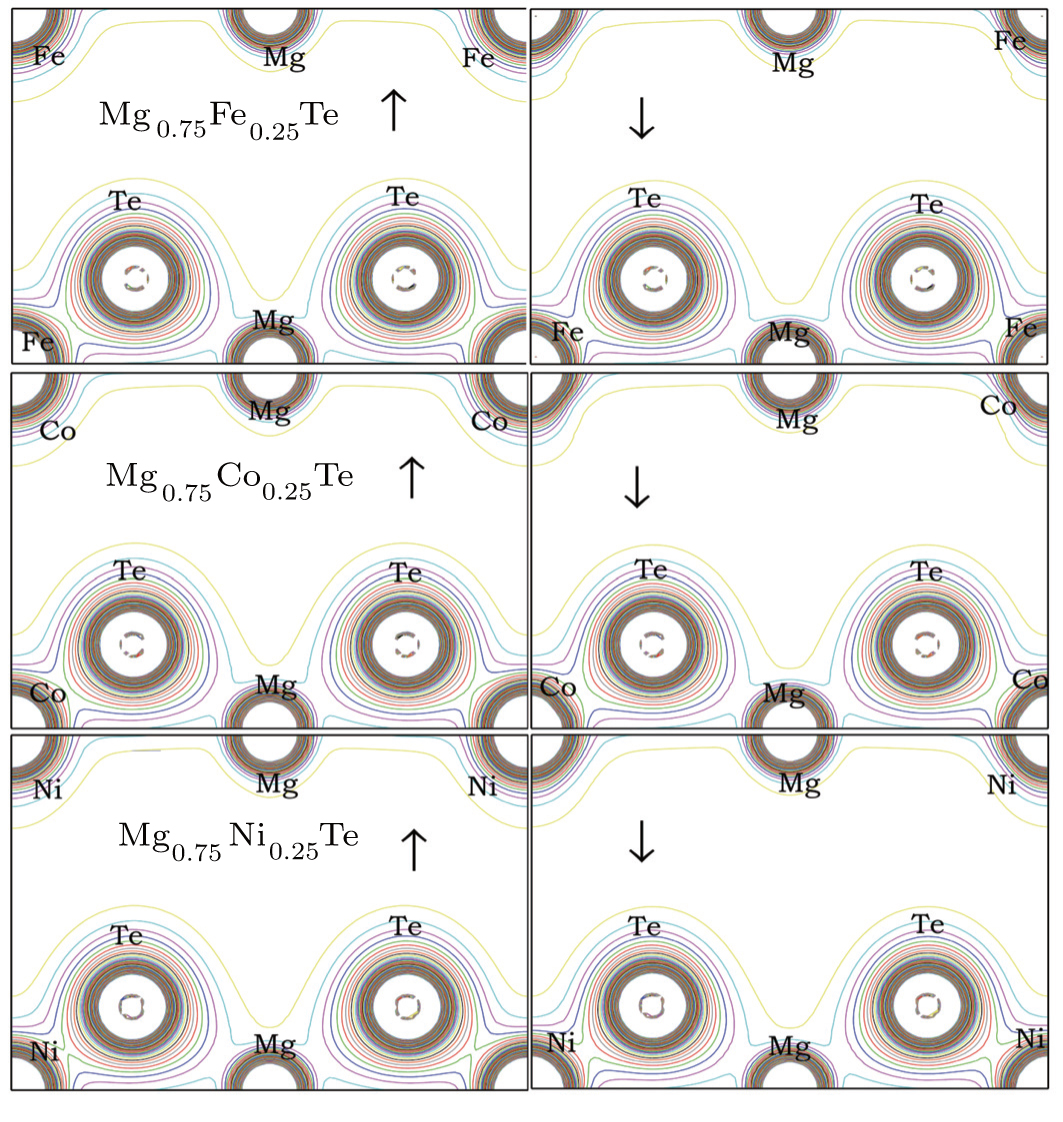
In order to understand the charge transfer properties of the spin-polarized Mg0.75TM0.25Te compounds and elaborate their bonding natures, we compute the valence charge densities of Mg, TM, and Te, which are shown in Fig. 6. It is evident from the plots of the charge density contour that the Mg0.75TM0.25Te compounds have mixed ionic and covalent character for the Mg–Te and TM–Te bonds. However, the Mg–Te bonds are more ionic in nature than TM–Te due to larger electronegativity difference between Mg and Te. The valence charge around the Mg site is due to s states, while the Te site shows both s and p valence charge states. The charge density appears to be the same at Mg and Te sites for the three TM dopants considered in this work, and their differences only appear at the TM site. The similar electronegativity values of the TM elements considered in the present study ensure that no major differences appear in their valence charge density plots except for the fact that the 3d-eg states appear above the Fermi level (and below CB minima) as the spin-down charge density varies at the TM site in the cases of Mg0.75Fe0.25Te and Mg0.75Co0.25Te. In contrast, the Mg0.75Ni0.25Te compound has almost similar charge density plots for both spin-up and spin-down cases, which can be attributed to less difference in energy positioning of its 3d-t2g and 3d-eg states than Fe- and Co-doped MgTe.
Fig. 6. Plots of spin polarized contour of (a) Mg0.75Fe0.25Te, (b) Mg0.75Co0.25Te, and (c) Mg0.75Ni0.25Te alloys, obtained by using the mBJ potential for majority spin (↑) and minority spin (↓).
3.3. Magnetic properties
In transition metal doped diluted magnetic semiconductors, the partially filled 3d state creates a major part of the magnetic moment through strong p–d interaction. The total and partial magnetic moments of Mg0.75TM0.25Te (TM = Fe, Co, Ni) compounds are shown in Table 3. The exchange mechanism between magnetic ions and charge carriers (electrons or holes) near the band edges provides distinct properties for magnetic semiconductors. The Zener free electron model, RKKY model and double exchange model describe the hybridizations (s-d, p–d, and d–d) to elaborate ferromagnetism in these alloys. Therefore, Zeners and double exchange interaction (the interaction between magnetic ions and free carriers of the nonmagnetic host semiconductor)[51] are responsible for ferromagnetism in Fe, Co, and Ni doped MgTe alloys. It is clear from the above discussion that the reduction of magnetic moment of transition metal ions and the generation of magnetic moment in the neighboring nonmagnetic ions are due to p–d hybridization between impurity (TM) d-band and Te p-band.
Table 3.
Table 3.
Table 3.
Calculated values of total and local magnetic moment (in Bohr magneton) for the FM Mg1−xMxTe (M = Fe, Co, and Ni) at x = 0.25 alloys within the muffin tin spheres and in the interstitial sites.
.
Compounds
Total/μB
Mg/μB
Te/μB
M = Fe, Co, and Ni/μB
interstitial/μB
Mg0.75Fe0.25Te
4.00022
0.03764
0.03764
3.44063
0.36274
Mg0.75Co0.25Te
3.00017
0.01033
0.03829
2.46935
0.34669
Mg0.75Ni0.25Te
2.00017
0.00873
0.07654
1.31347
0.35435
Table 3.
Calculated values of total and local magnetic moment (in Bohr magneton) for the FM Mg1−xMxTe (M = Fe, Co, and Ni) at x = 0.25 alloys within the muffin tin spheres and in the interstitial sites.
.
Referring to Figs. 3–5 and Table 3, overlapping of TM 3d states with Te p states near the Fermi level is significant, especially in a range of 1 eV–5 eV. In all of these causes, the TM-d states interact with the Te-p states strongly, resulting in the splitting of the energy levels which are closer to the Fermi level. Consequently, the Te octahedral crystal field splits the TM 3d state (five-fold degenerate) into the two-fold degenerate high-lying 3d-eg (dz2 and dx2 − y2) and the three-fold degenerate low-lying 3d-t2g (dxy, dxz, and dyz) symmetry states. The high energy of 3d-eg states as compared with that of 3d-t2g states indicates that the TM ion occupies the octahedral surroundings.
Figure 7 shows the spin-dependent density along (110) planes as an overall effect of electron spins (combined effect of spin up and spin down states). It is clear from Fig. 7 that a major part of the magnetic moment comes from transition metal ions with small contributions of Mg and Te, which is also confirmed from Table 4. Moreover, the values of spin exchange splitting energy Δx (d)[52] that explains the contribution of transition metal 3d state in exchange mechanism are calculated for Fe-, Co-, and Ni-doped MgTe, and their numerical values are 3.30 eV, 2.90 eV, and 2.82 eV, respectively. Moreover, the values of crystal field energy that determines the difference in energy gap between anti-bonding and bonding state of 3d splitting are 0.65 eV, 0.52 eV, and 0.40 eV respectively. The comparison of these two energies shows that the spin exchange splitting energy is greater than the crystal field splitting energy for each concentration because the bonding state does not contribute to the exchange mechanism, which favors the ferromagnetism.
Fig. 7. Plots of spin density for (a) Mg0.75Fe0.25Te, (b) Mg0.75Co0.25Te, and (c) Mg0.75Ni0.25Te alloys, with setting the iso-value to be 1 × 10−5.
Table 4.
Table 4.
Table 4.
Calculated values of band gap values, conduction and valance band edge splitting and exchange constants (N0α & N0β) in unit eV for FM Mg1−xMxTe (M = Fe, Co, and Ni) at x = 0.25 alloys.
Calculated values of band gap values, conduction and valance band edge splitting and exchange constants (N0α & N0β) in unit eV for FM Mg1−xMxTe (M = Fe, Co, and Ni) at x = 0.25 alloys.
.
The most prominent parameters calculated from s–d and p–d coupling are exchange constants N0α and Nβ, respectively,[53] which determine the exchange interaction between TM d-state and charge carriers (electrons in the conduction band and holes in the valance band). The values of exchange constants for Mg0.75Fe0.25Te, Mg0.75Co0.25Te, and Mg0.75Ni0.25Te are presented in Table 4. The negative values of both parameters confirm parallel coupling (double exchange mechanism). Further, N0α has a much lower value than Nβ which means that the s–d interaction at the conduction band minimum is much weaker than the p–d interaction at valance band maximum, which indicates the ferromagnetic behaviors expected in these alloys.
4. Conclusions
We systematically evaluate the structural, elastic, electronic and magnetic properties of Mg0.75TM0.25Te (TM = Fe, Co, Ni) magnetic semiconductors by the ab-initio method. The structures of Mg0.75TM0.25Te (TM = Fe, Co, Ni) compounds are optimized in PM, FM, and AFM states. The positive values of total energy differences (ΔE1 and ΔE2) show that our alloy is stable in the FM phase. Our predicted results of lattice parameter and bulk modulus are reported for the first time, which seem to be reliable as the estimated values of bulk modulus using the relation B = (1/3) (C11+2C12) and by the energy minimization method have nearly the same values. The estimated values of elastic parameters ν and B/G show that the doping of TM can change from the brittle to the ductile nature. The band structure reveals that each of all the compounds has a direct band gap in the spin-up channel and the calculated values of bandgap for Mg0.75TM0.25Te (TM = Fe, Co, Ni) are smaller than for the parent compound MgTe. The reduction of magnetic moment of TM ions and the generation of magnetic moment in the neighboring nonmagnetic ions can be justified by p–d hybridization and may also be verified from the spin density plots. The greater values of crystal field energy than exchange splitting energy indicate that these compounds each possess ferromagnetism. The lower value of N0α than Nβ shows that that p-d interaction at valance band maximum in the spin-down state is more effective and their strong hybridization confirms the ferromagnetism.
BlahaPSchwarzKMadsenG K HKvasnickaDLuitzJ2001WIEN2K — An augmented-plane-wave+local orbitals program for calculating crystal propertiesKarlheinz Schwarz, Techn.Wien, Austria2001, ISBN 3-9501031-1-2
Investigations of mechanical, electronic, and magnetic properties of non-magnetic MgTe and ferro-magnetic Mg0.75TM0.25Te (TM = Fe, Co, Ni): An ab-initio calculation
[Mahmood Q1, Alay-e-Abbas S M2, 3, Mahmood I4, Asif Mahmood5, Noor N A4, †, ]







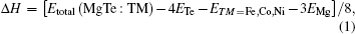
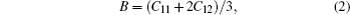



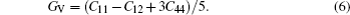

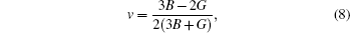



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 ]
]