
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Unifying the crystallization behavior of hexagonal and square crystals with the phase-field-crystal model
[Yang Tao†,  , Chen Zheng, Zhang Jing, Wang Yongxin, Lu Yanli]
, Chen Zheng, Zhang Jing, Wang Yongxin, Lu Yanli]
, Chen Zheng, Zhang Jing, Wang Yongxin, Lu Yanli]
|
|


† Corresponding author. E-mail:
Project supported by the National Natural Science Foundation of China (Grant Nos. 54175378, 51474176, and 51274167), the Natural Science Foundation of Shaanxi Province, China (Grant No. 2014JM7261), and the Doctoral Foundation Program of Ministry of China (Grant No. 20136102120021).
By employing the phase-field-crystal models, the atomic crystallization process of hexagonal and square crystals is investigated with the emphasis on the growth mechanism and morphological change. A unified regime describing the crystallization behavior of both crystals is obtained with the thermodynamic driving force varying. By increasing the driving force, both crystals (in the steady-state) transform from a faceted polygon to an apex-bulged polygon, and then into a symmetric dendrite. For the faceted polygon, the interface advances by a layer-by-layer (LL) mode while for the apex-bulged polygonal and the dendritic crystals, it first adopts the LL mode and then transits into the multi-layer (ML) mode in the later stage. In particular, a shift of the nucleation sites from the face center to the area around the crystal tips is detected in the early growth stage of both crystals and is rationalized in terms of the relation between the crystal size and the driving force distribution. Finally, a parameter characterizing the complex shape change of square crystal is introduced.
Remarkable diversity of growth patterns during crystallization has drawn long-standing attention of material scientists.[1,2] Crystal morphology varies greatly with the ordering condition changing, encompassing the faceted structures to symmetric dendrites, which in turn produces an equally diverse range of physical properties. Understanding of this diversity requires the considerations from both interfacial dynamics and external fields.[3–6] Interfacial dynamics determined by the interplay between interface energy anisotropy and interface kinetic anisotropy governs the complex growth behavior of crystal, while the external fields exert a large influence on the variation of interface anisotropies. Relying on the difference of interface absorption ability, Siegfried et al. have succeeded in controlling the morphology evolution of cuprous oxide by altering specified interfacial energies.[7] Through the tuning of supersaturation, Xu et al. discovered the dependence of growth pattern and induction time of KADP on ammonium concentration.[8]
Due to the complexities in experiments to dynamically capture the crystallizing process in atomic length scale, computer simulation has been proven to be a powerful tool in this regard.[9–17] As an extension of dynamical density functional theory,[15–17] the phase-field-crystal (PFC) model is capable of addressing the crystallization kinetics at atomic level while operating on diffusive timescale. Moreover, the crystalline anisotropy and growth driving force are naturally integrated in the PFC model by adopting an orientation-invariant free energy functional. Called for by the increasing importance of two-dimensional (2D) materials, a systematical study of revealing the crystallization mechanism in 2D materials, particularly for the hexagonal lattice and square lattice, is performed. In this work, by checking the atomic growth process using the PFC model, we focus on the interplays among the growth morphology, thermodynamic driving force, and the crystal structure, and resort to finding a unified description of crystallization behavior.
Based on the time-averaged atomic density, the dimensionless form of PFC free energy functional is constructed as[15,16]
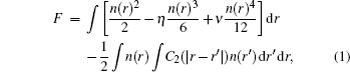
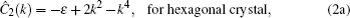
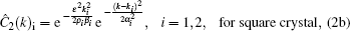
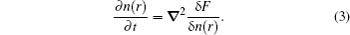
In order to stabilize respective structures, simulation parameters for the hexagonal square are set to be (η, v) = (0, 3) and ε = –0.6, while for square crystal (η, v) = (1, 1), ε = 0, 
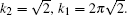
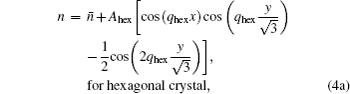
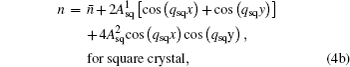
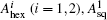
In the PFC model, the driving force of crystallization can be altered by changing the supersaturation of the system (here defined as σ =(n̄ – n̄liq)/(n̄sq – n̄liq),[17] where n̄liq and n̄sq represent the average densities of the equilibrium liquid and solid phases, respectively), i.e., by changing the initial average density of the liquid n̄. In this section, we will first present the crystallization process of hexagonal crystal with the average density n̄ ranging from −0.49 to −0.462, and then, followed by the description of the growth process of square crystal with the average density varying from n̄ = −0.1 to − 0.05. In the end, we will relate our simulation results to the experiments.
As shown in Fig. 


Figure 
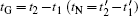
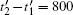
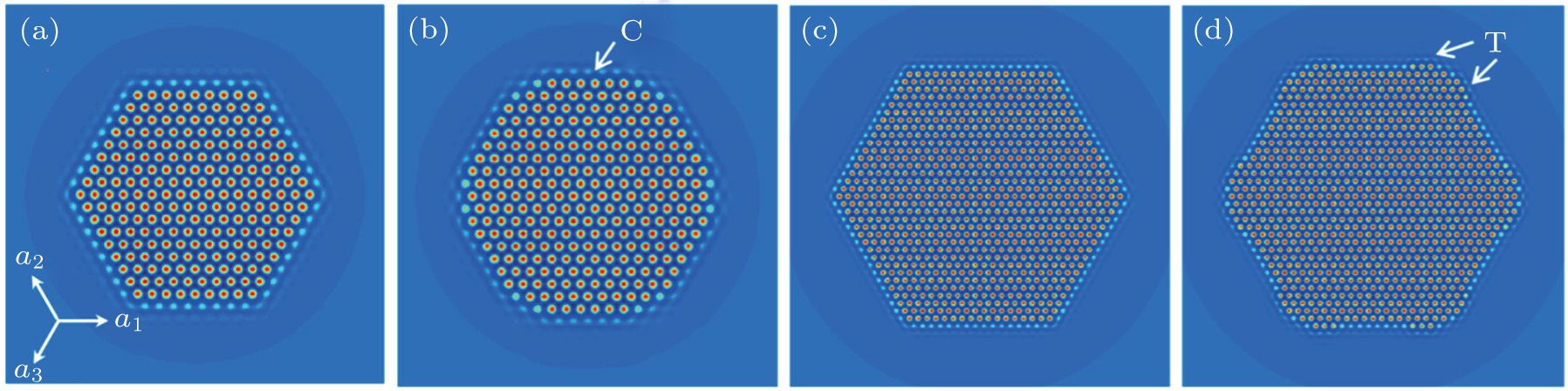 | Fig. 1. Crystallization process of hexagonal crystal structure simulated with n̄ = −0.49 at different times: (a) t = 3500, (b) t = 4000, (c) t = 18500, and (d) t = 19000. For a better visualization, magnification factors of panels (a) and (b) are larger than those of panels (c) and (d). |
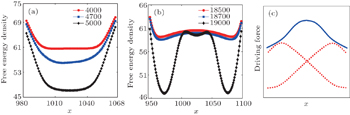
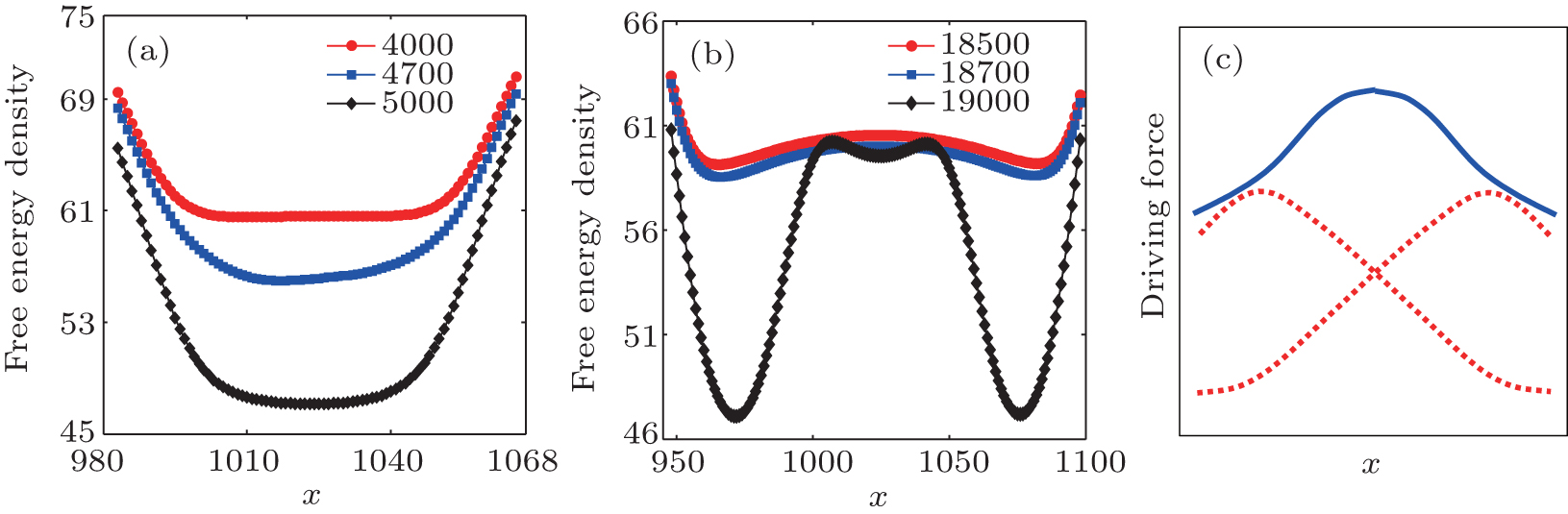 | Fig. 2. (a) and (b) The variation of free energy density along |
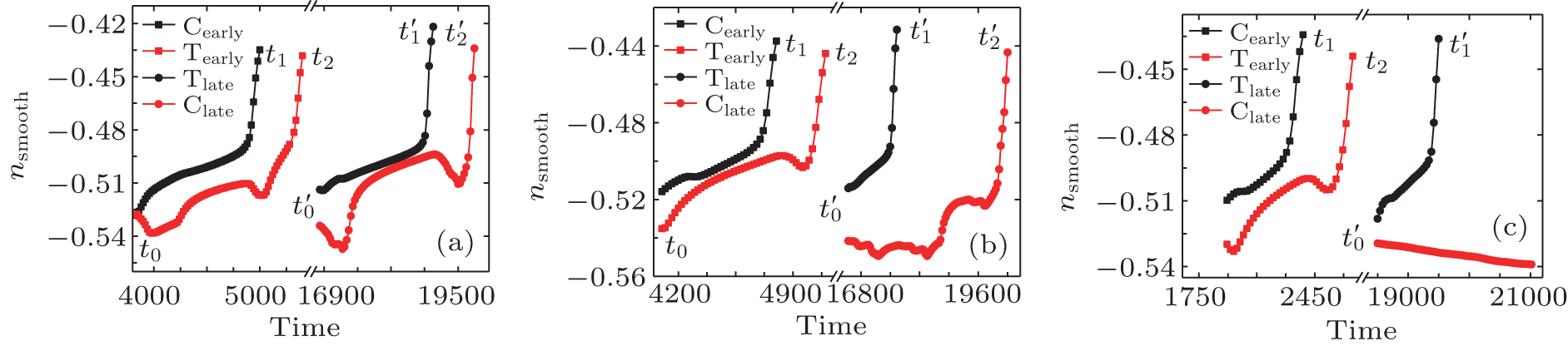 | Fig. 3. The variation of smoothed atomic density at positions C and T (shown in Fig. |
Figure 
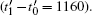
Based on the above discussion, it is shown that the interface migration mechanism and crystal shape are closely related to the thermodynamic driving force. With the driving force increasing, the period of nucleation drops faster than that of growth, undermining the stability of LL growth mode and favoring the ML growth mode. Thus, the ML growth mode is more favored as the average density n̄ is raised up. By setting the parameter n̄ = −0.465, the crystal initially shows as a faceted hexagon, then the edges concave in, and finally the crystal develops into a dendrite, as shown in Figs. 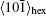
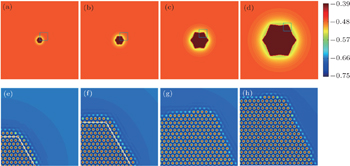
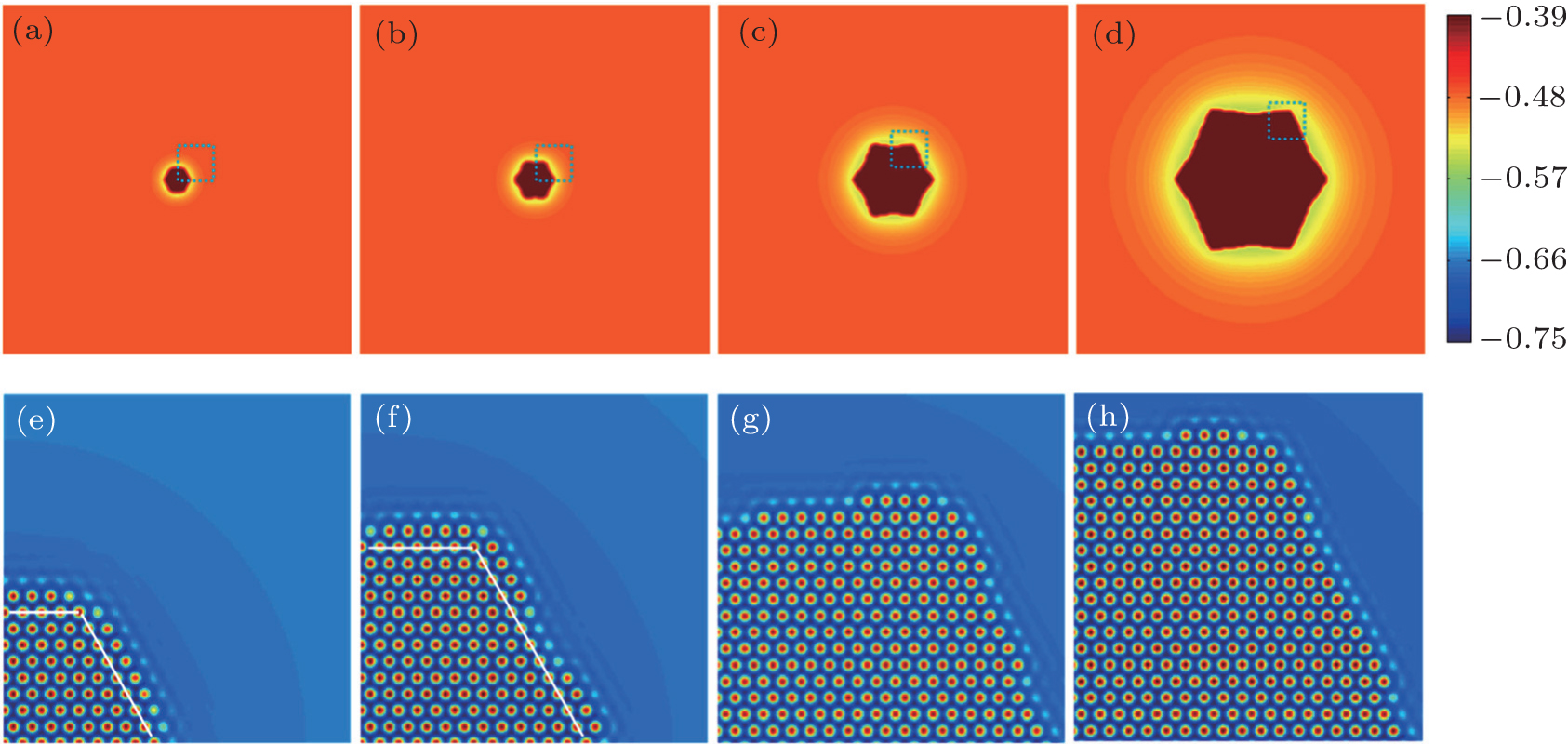 | Fig. 4. Growth morphology of hexagonal crystal and the corresponding atomic density fields for the parts enclosed by dashed boxes, simulated with parameter n̄ = −0.47 at different times: (a) and (e) t = 2100, (b) and (f) t = 5000, (c) and (g) t = 19200, and (d) and (h) t = 72000. The white lines in panels (e) and (f) outline the newly generated layers. The color bar in the top row represents the variation of the smoothed atomic density field nsmooth. |
 | Fig. 5. Growth morphology of hexagonal crystal and the corresponding atomic density maps for the parts enclosed by dashed boxes simulated with parameter n̄ = −0.462 at different times: (a) and (e) t = 1600, (b) and (f) t = 4000, (c) and (g) t = 20000, and (d) and (h) t = 63000. The white lines in panels (e) and (f) outline the newly generated layers. The color bar in the top row represents the variation of the smoothed atomic density field nsmooth. |
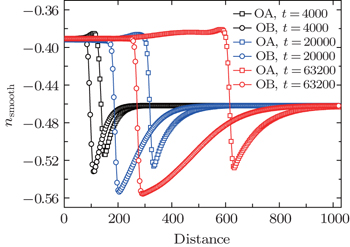
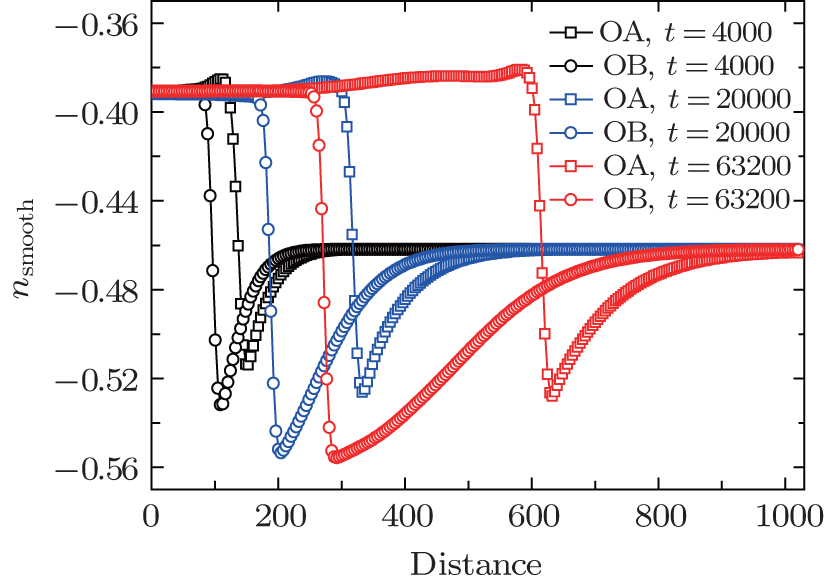 | Fig. 6. The smoothed density profiles of hexagonal crystal along lines OA and OB (shown in Fig. |
Similar growth patterns and migration mechanism are also found in the crystallization process of square crystal when increasing the driving force, as shown in Fig.
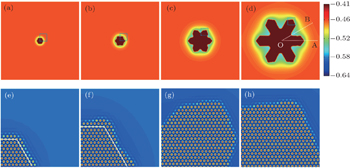
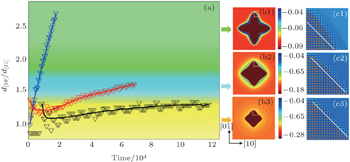
 | Fig. 7. (a) Distance ratios of growth fronts along [10]sq and [11]sq directions, (b1)–(b3) three characteristic morphologies and (c1)–(c3) the corresponding atomic configurations of the area enclosed by the white box in panels (b1)–(b3). Black, red, and blue triangle symbols in panel (a) are simulated with parameter n̄ = −0.1, −0.07, and −0.05, and the curves represent the corresponding smoothed data. The color bars in panels (b1)–(b3) mark the variation of the smoothed atomic density field nsmooth. |
Our simulations reveal the orientation selection process associated with various growth patterns of hexagonal crystal and square crystal, which is helpful to illuminate the physical origins of morphological diversity. In our simulations, a non-uniform distribution of driving force around the crystal causing a preferred nucleation at positions near the corners is observed, in conformity with the Berg effect. On the other hand, in experiments during the growth of colloidal films and isotactic polystyrene,[19,20] the crystal displays as a faceted hexagon at low driving force, and transforms to the edge-concave morphology at intermediate driving force and then to the six-fold dendrite when further increasing the driving force, which is in good accordance with the morphological change depicted in Figs.
In conclusion, the morphological transition and growth mechanism of hexagonal and square crystals under the variation of thermodynamic driving forces are investigated at the atomistic scale using the PFC model. The interface of hexagonal crystal advances through the nucleation of
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 | |
| 22 |