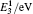
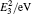
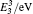
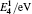
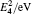
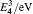
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Boron diffusion in bcc-Fe studied by first-principles calculations
[Li Xianglong1, Wu Ping1, †,  , Yang Ruijie1, Yan Dan1, Chen Sen1, Zhang Shiping1, Chen Ning2]
, Yang Ruijie1, Yan Dan1, Chen Sen1, Zhang Shiping1, Chen Ning2]
, Yang Ruijie1, Yan Dan1, Chen Sen1, Zhang Shiping1, Chen Ning2]
|
|


† Corresponding author. E-mail:
Project supported by the National Natural Science Foundation of China (Grant No. 51276016) and the National Basic Research Program of China (Grant No. 2012CB720406).
The diffusion mechanism of boron in bcc-Fe has been studied by first-principles calculations. The diffusion coefficients of the interstitial mechanism, the B–monovacancy complex mechanism, and the B–divacancy complex mechanism have been calculated. The calculated diffusion coefficient of the interstitial mechanism is D0 = 1.05 × 10−7 exp (−0.75 eV/kT) m2 · s−1, while the diffusion coefficients of the B–monovacancy and the B–divacancy complex mechanisms are D1 = 1.22 × 10−6 f1 exp (−2.27 eV/kT) m2 · s−1 and D2 ≈ 8.36 × 10−6 exp (−4.81 eV/kT) m2 · s−1, respectively. The results indicate that the dominant diffusion mechanism in bcc-Fe is the interstitial mechanism through an octahedral interstitial site instead of the complex mechanism. The calculated diffusion coefficient is in accordance with the reported experiment results measured in Fe–3%Si–B alloy (bcc structure). Since the non-equilibrium segregation of boron is based on the diffusion of the complexes as suggested by the theory, our calculation reasonably explains why the non-equilibrium segregation of boron is not observed in bcc-Fe in experiments.
It has been reported that the addition of trace amounts of boron can significantly improve the strength, hot ductility, and hardenability of ultra-low carbon microalloyed steel.[1,2] The beneficial effects may be related to the boron diffusion at high temperature and the boron segregation during the cooling process. Due to the experiment difficulties in detecting trace amounts of B (light element), the mechanism of the diffusion and segregation of boron still need to be investigated.
The non-equilibrium segregation of boron has drawn many researchers’ attention and a theory has been proposed.[3,4] In the theory, it is assumed that, in the grains, there is a certain concentration of boron–vacancy complexes. When the sample is quenched at high temperature, the supersaturated vacancies and complexes may diffuse to the grain boundaries (sink of vacancy). After the annihilation of the vacancies, the surplus boron atom will stay at the boundaries and the segregation occurs. According to Wu et al.,[5] the non-equilibrium segregation of boron is mainly observed in the steel cooled down from the fcc structure. Zhang et al.[6] did not detect non-equilibrium segregation of boron in bcc Fe–3%Si–B alloy by the particle-tracking autoradiograph (PTA) method. Therefore, it may be meaningful to study the diffusion mechanism of boron in the bcc structure.
Generally, a solute atom at a substitutional site may diffuse with a solute–vacancy mechanism, while that in an interstitial site may diffuse with an interstitial mechanism.[7] The experiments of Busby et al.[8] and Wang et al.[9] showed that boron tends to occupy a substitutional site, however, the calculations of Bialon et al.[10] and Fors et al.[11] showed that boron diffuses with an interstitial mechanism. Since the viewpoints on the diffusion mechanism are inconsistent with the solution type, a more detailed study on the diffusion mechanism of boron is needed.
Divacancies are more mobile than monovacancies, and can be more effective diffusion vehicles than monovacancies.[7] In the study of the self-diffusion of sodium (bcc structure) at high temperature, Ho[12] found that the divacancy mediated diffusion plays an important role in the diffusion of sodium. To clarify the diffusion mechanism of boron, both the contributions of the monovacancy and the divacancy should be taken into consideration.
Recently, the first-principles calculations based on the density functional theory (DFT) have been widely applied to study the interactions and the complex of point defects in crystals.[13] Taiquan[14] calculated the interaction of a Ge atom with the vacancy in silicon single crystal and found the stable complexes. Liu et al.[15] investigated the diffusion behaviors of H isotopes in W, and calculated the diffusion factor of H, which is in agreement with the experimental value. Zhou et al.[16] studied the carbon trapping mechanism in copper, and found that a monovacancy is capable of trapping as many as four C atoms to form a CnV complex. Domain et al.[17] calculated the interactions between a C or N atom and a vacancy or other interstitials in α-Fe, and found that a vacancy can trap up to two C atoms. Ohnuma et al.[18] studied the interaction between solute atoms (C/N etc.) and the vacancy in bcc Fe, and obtained the binding energy and the stable configuration of the solute–vacancy complex.
In this work, the diffusion mechanism of boron in bcc-Fe is studied by first-principles calculations. The formation energies of boron at different sites and the binding energies between the point defects are calculated in detail. Then, the diffusion coefficients of the interstitial mechanism and the complex mechanism are calculated and the diffusion mechanism is analyzed.
All the calculations were performed by using the CASTEP code.[19] The electronic exchange–correlation terms were described with the generalized gradient approximation (GGA) proposed by Perdew et al. (PW91).[20] Ultrasoft pseudo-potentials for all the elements were used. The cutoff energy of the atomic wave functions was fixed at 450 eV for all calculations to achieve a good convergence. The calculations were performed using 3×3×3 supercells which are periodic in all three directions. The energy integration over a Brillouin zone was performed with 4×4×4 k-points grids according to Monkhorst–Pack.[21] The spin-polarized approximation was adopted and a ferromagnetic model was used.
The formation energy of single point defect i at a substitutional (S) site 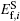
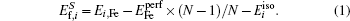
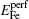
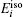
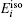
The formation energy of an interstitial boron atom 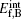
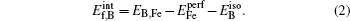
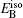
The binding energy of two defects Eb (i, j) is defined as
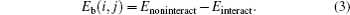
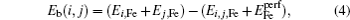
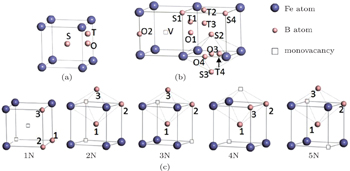
The configurations with the defects we are concerned with are shown in Fig.
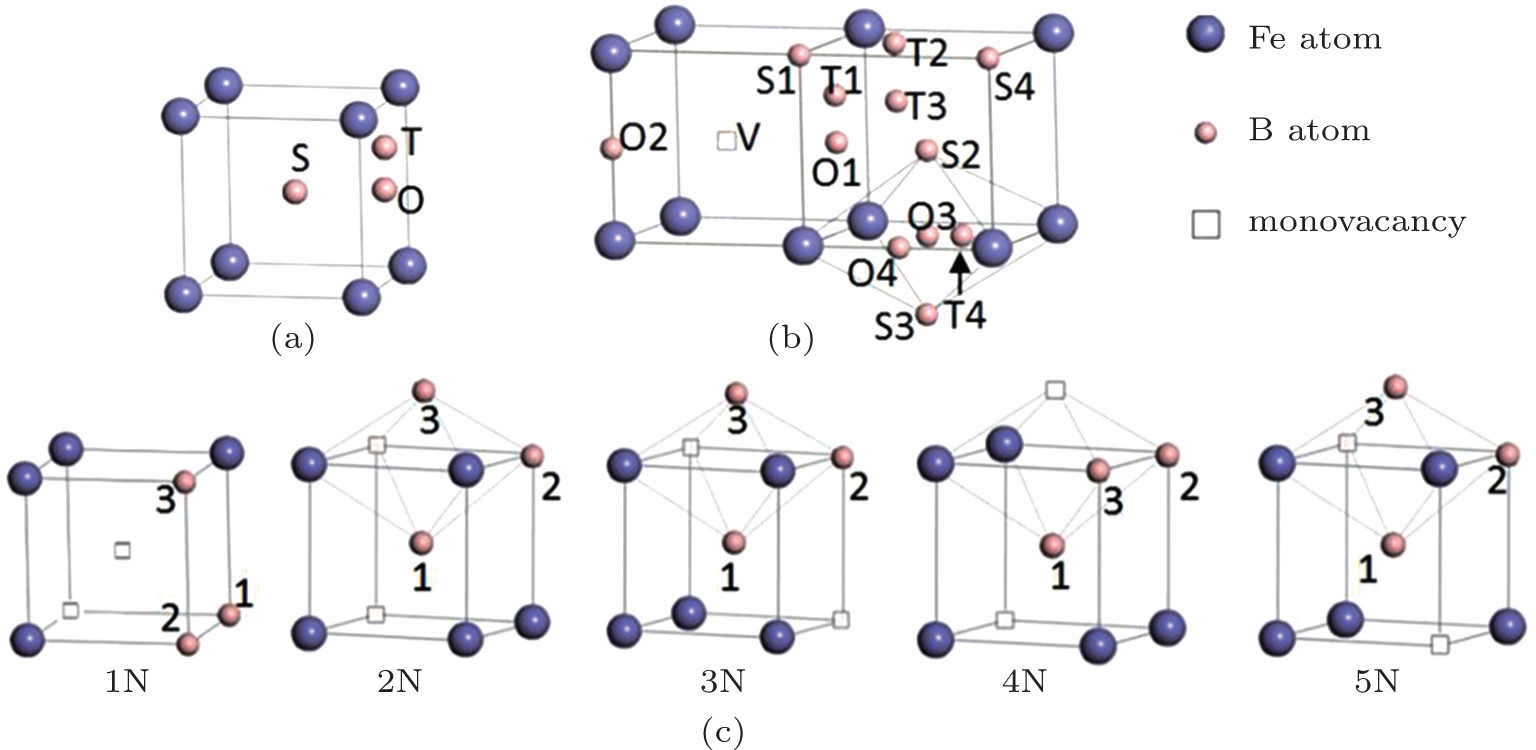 | Fig. 1. The configurations used in this work. (a) Boron atom at the substitutional site (S), the tetrahedral interstitial site (T), and the octahedral interstitial site (O). (b) The configurations of a boron atom and a monovacancy. Around the monovacancy (V), the first four (1, 2, 3, and 4) nearest S, O, T sites of the boron atom are shown. (c) The configurations of a divacancy and an adjacent boron atom near the divacancy. The first five nearest neighboring divacancies (1N, 2N, 3N, 4N, 5N) with boron atoms at their first three nearest substitutional sites (1, 2, and 3) are shown. |
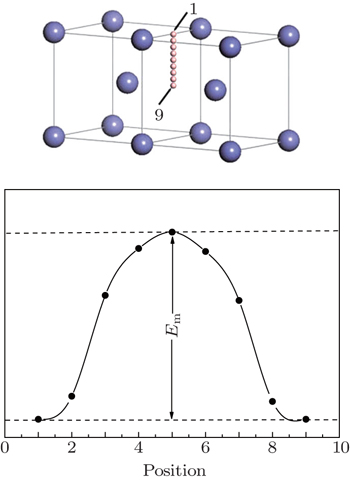
The diffusion coefficient of boron diffusion through octahedral interstitial sites is
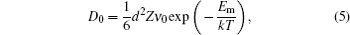
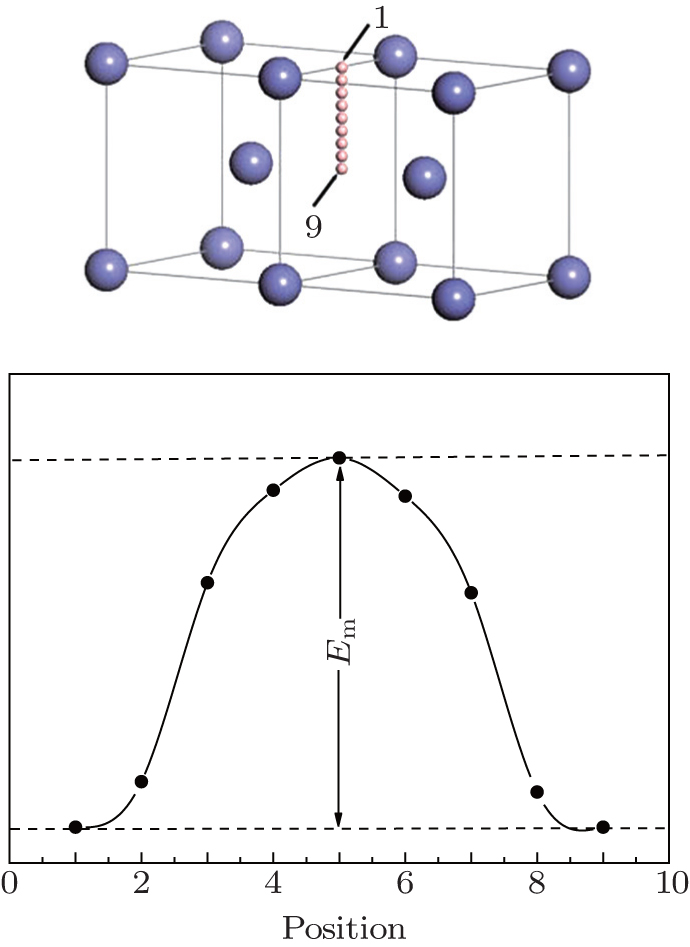 | Fig. 2. (a) The jumping atom was placed at the evenly spaced sites along the jumping path in sequence. Then, the supercell was fully optimized with the coordinates of the jumping atom fixed, and the energy of the supercell was calculated. (b) Finally, the jumping barrier was obtained from the results. |
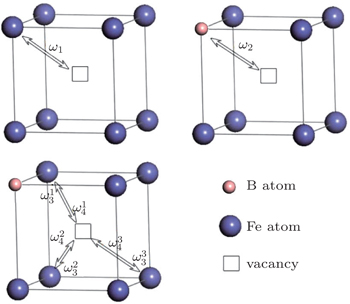
For the complex mechanisms, the solute atom diffuses through a series of exchanges with neighbor vacancies, and the diffusion coefficient is related to the exchange frequencies, the correlation factor, and the concentration of vacancies. The correlation factor is a quantity introduced so that the successive jumps (exchanges) are correlated, and a larger correlation factor means a lower probability to jump backward. In this work, two mechanisms were taken into consideration, which are the boron–monovacancy complex mechanism and the boron–divacancy complex mechanism.
The diffusion coefficient of the boron–monovacancy complex mechanism D1 is[7]
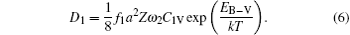
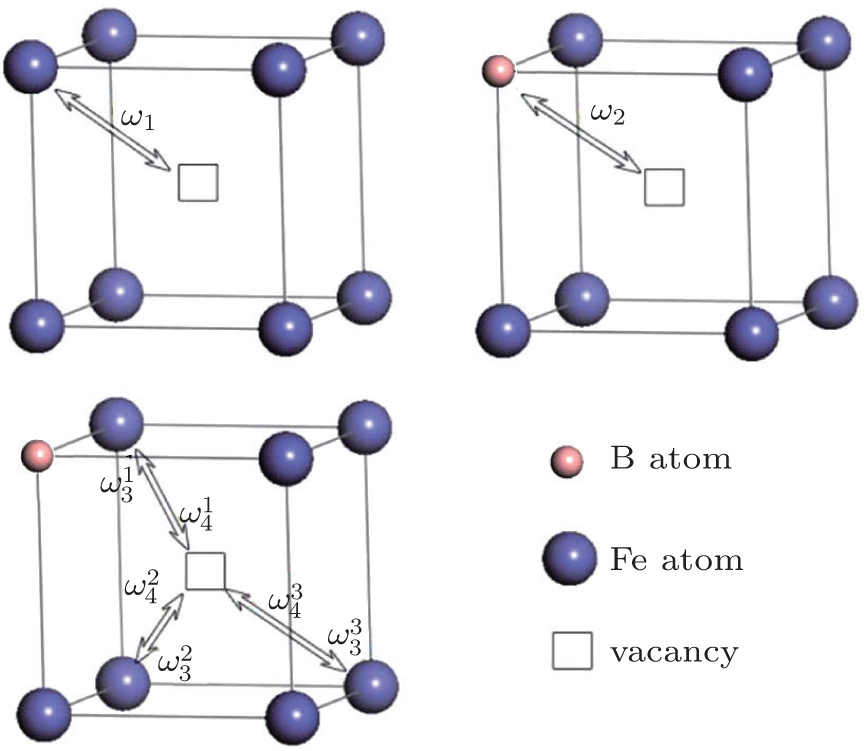 | Fig. 3. The four-frequency model of boron diffusion in bcc-Fe. Here, ω1 is the solvent-vacancy exchange frequency, ω2 is the solute–vacancy exchange frequency, ω3 is the dissociation frequency of the complex, and ω4 is the association frequency of the complex. |
According to the four-frequency model of Manning,[22] the correlation factor f1 can be calculated as
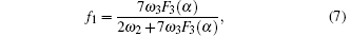
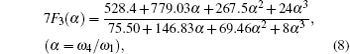
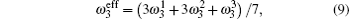
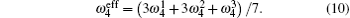
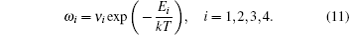
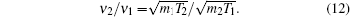
The concentration of the monovacancy C1V in Eq. (
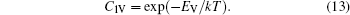
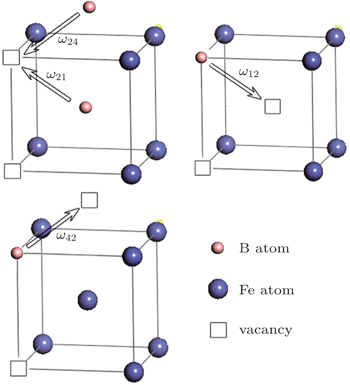
The diffusion mechanism of the divacancy in the bcc structure was proposed by Mehrer.[25] The migration of a divacancy involves three configurations (1N, 2N, 4N), which are shown in Fig.
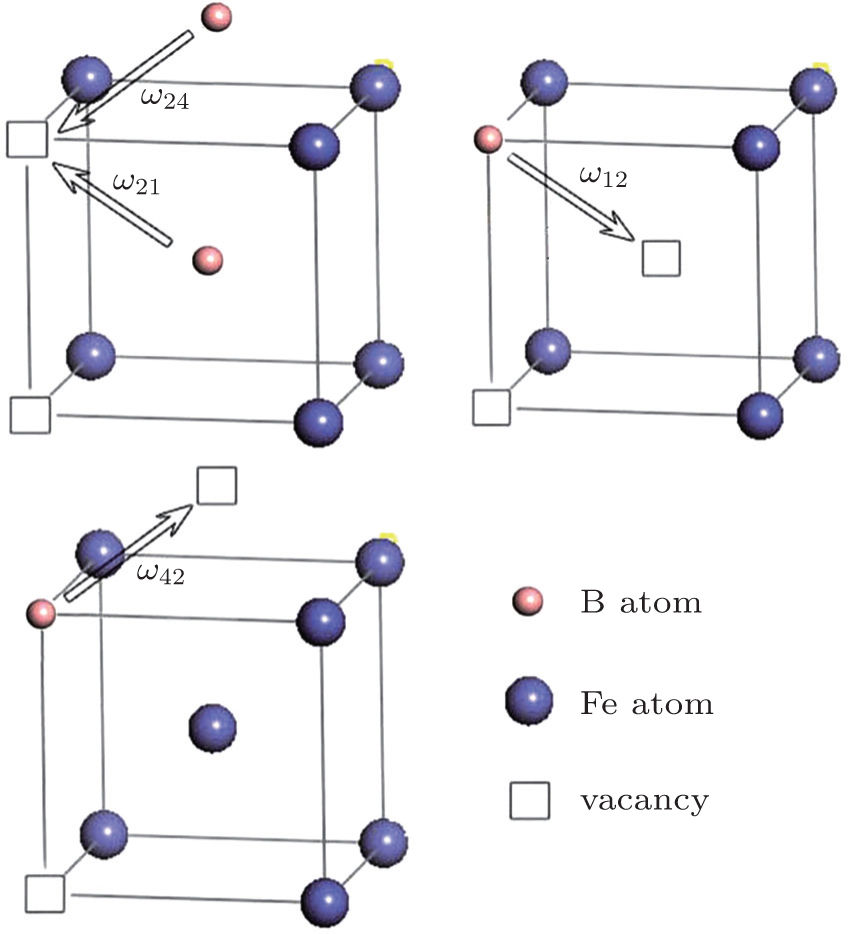 | Fig. 4. The divacancy configurations involved in the migration and the exchange frequencies between divacancies: 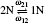 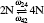 |
The diffusion coefficient of the boron–divacancy complex mechanism can be calculated with the method proposed by Mehrer[25]
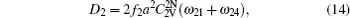
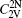
The concentration of the divacancy 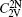
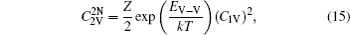
The ω21 and ω24 can be calculated by the method introduced in Eq. (
The correlation factor f2 can be calculated with the method proposed by Belova et al.[26]
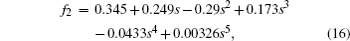
The formation energies of the point defects were calculated. A boron atom at a substitutional site (S), an octahedral site (O), and a tetrahedral site (T) was introduced into the supercell respectively. The configurations are shown in Fig.
| Table 1. The formation energy of a monovacancy (V) and a boron atom at a substitutional site (S), an octahedral interstitial site (O), and a tetrahedral interstitial site (T). . |
The results show that the formation energy of the vacancy is higher than that obtained by Domain et al.[27] The formation energy of the boron atom at O site (EO) is higher than that at S site (ES), while the formation energy of the boron atom at T site (ET) is much higher, and these are in accordance with that obtained by Fors et al.[11] Hence, we conclude that the boron atom tends to occupy the S or O site instead of the T site.
The binding energies of a boron atom at different sites (S, O, and T) with its neighbor monovacancy (V) have also been calculated. The configurations are shown in Fig.
| Table 2. The binding energy (in eV) of a boron atom (at S/O/T site) with a monovacancy at the first (1), the second (2), the third (3), and the fourth (4) nearest neighbor sites. . |
The binding energy of a boron atom at the S site with the monovacancy at the first and the second nearest neighbor sites (S–V1 and S–V2) are 0.25 eV and 0.21 eV, and they are higher than the others. The calculation of Bialon et al.[10] also showed that the S–V1 and S–V2 configurations are more stable.
In Table
It seems that the configuration becomes unstable when there is a monovacancy near the interstitial boron atom. Bialon et al.[10] also found that the configurations of O–V1 and O–V2 are unstable.
The binding energies of the first five nearest neighboring divacancies were calculated and the configurations are shown in Fig.
| Table 3. The binding energies (in eV) of the divacancies. . |
The results show that the binding energies of the 1N, 2N, and 4N nearest neighboring divacancies are larger than the others, and the binding energy of the 2N divacancy is the largest, which is in accordance with the results in the references.
The interaction between the boron atom and the neighboring divacancy was studied. As the configuration becomes unstable when there is a vacancy near the interstitial boron atom, only the boron atom at a substitutional site was considered. The binding energies between the five types of divacancies (1N, 2N, 3N, 4N, 5N) and the substitutional boron atom at their first three nearest neighboring sites (1nn, 2nn, 3nn) were calculated. The configurations used in the calculations are shown in Fig.
| Table 4. The binding energy between the substitutional atom and its neighboring divacancy. . |
The results show that there are four complexes of the substitutional boron atom and the divacancy, whose binding energies are higher. They are the 1N, 2N, and 4N divacancies with the boron atom at the first nearest site (1N–1nn, 2N–1nn, 4N–1nn), and the 3N divacancy with the boron atom at the second nearest site (3N–2nn).
According to Mehrer,[25] there are three different configurations involved in the migration of a divacancy in the bcc structure, which are the 1N, 2N, and 4N divacancies.
In our results, the binding energies of the 1N/2N/4N divacancies and the 1nn boron atom are larger than most of the others, which means that they are likely to form complexes. Therefore, it is necessary to calculate the effect of the divacancy in the diffusion of the boron atom.
In this section, the diffusion coefficients of both the interstitial mechanism and the complex mechanism are studied.
The diffusion coefficient can be calculated with Eq. (
| Table 5. The parameters for the calculation of the diffusion coefficient. . |
As shown in Fig.
The jumping barrier energy and the jumping frequency needed for the calculation of the diffusion coefficient are listed in Table
| Table 6. The parameters needed for the calculation of the diffusion coefficient. . |
The correlation factor f1 can be calculated with the methods introduced before and the results are shown in Table
| Table 7. The correlation factors of the boron–monovacancy mechanism (f1). . |
The correlation factors of the boron–monovacancy diffusion mechanism at different temperatures are shown in Table
The diffusion coefficient of the boron–monovacancy complex mechanism can be obtained from Eq. (
The parameters for the calculation of the diffusion coefficient of the boron–divacancy complex are shown in Table
| Table 8. The parameters needed for the calculation of the diffusion coefficient. . |
The correlation factor of the boron–divacancy mechanism has been calculated with Eq. (
| Table 9. The correlation factors of the boron–divacancy mechanism (f2). . |
Thus, the diffusion coefficient of the boron–divacancy complex mechanism can be deduced from Eq. (
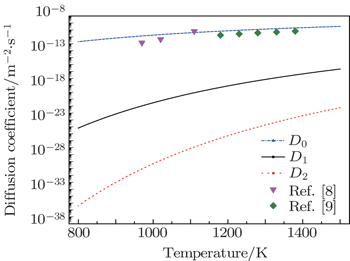
The diffusion coefficients of the interstitial mechanism and the boron–vacancy mechanism are shown in Fig.
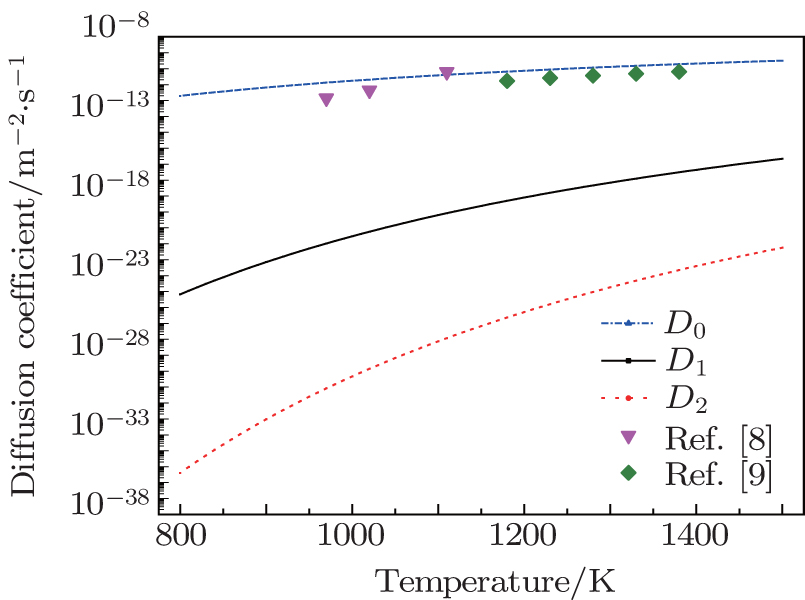 | Fig. 5. The calculated diffusion coefficients of different diffusion mechanisms and the results from experiments. |
Our calculations together with the calculation of Bialon et al.[25] show that the interstitial solute atom may enter a substitutional site when there is a vacancy near it. However, as mentioned above, the high formation energy of a vacancy makes the concentration of the vacancies too low to have an effect on the interstitial diffusion mechanism. Therefore, the dominant diffusion mechanism in boron is still the interstitial diffusion mechanism.
The experiment results of Busby et al.[8] and Wang et al.[9] are shown in Fig.
Our results show that the dominant diffusion mechanism is the interstitial mechanism. According to the theory of non-equilibrium segregation, the non-equilibrium segregation cannot form without the diffusion of the boron–vacancy complex. Zhang et al.[6] have studied the segregation of boron in bcc-Fe by the PTA method, no observable non-equilibrium segregation was detected in bcc-Fe, and this agrees with our conclusion.
The boron diffusion coefficients of the interstitial mechanism and the complex mechanism in bcc-Fe have been obtained by first-principles calculations. In particular, for the complex mechanism, both the monovacancy and the divacancy mechanisms were considered. The results are summarized as follows.
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 | |
| 22 | |
| 23 | |
| 24 | |
| 25 | |
| 26 | |
| 27 | |
| 28 |