
{kind=link}
Ab initio investigation of sulfur monofluoride and its singly charged cation and anion in their ground electronic state
[Li Song†,  , Chen Shan-Jun, Chen Yan, Chen Peng]
, Chen Shan-Jun, Chen Yan, Chen Peng]
, Chen Shan-Jun, Chen Yan, Chen Peng]
|
|


† Corresponding author. E-mail:
Project supported by the National Natural Science Foundation of China (Grant Nos. 11304023 and 11447172), the Young and Middle-Aged Talent of Education Burea of Hubei Province, China (Grant No. Q20151307), and the Yangtze Youth Talents Fund of Yangtze University, China (Grant No. 2015cqr21).
The SF radical and its singly charged cation and anion, SF+ and SF−, have been investigated on the MRCI/aug-cc-pVXZ (X = Q, 5, 6) levels of theory with Davidson correction. Both the core–valence correlation and the relativistic effect are considered. The extrapolating to the complete basis set (CBS) limit is adopted to remove the basis set truncation error. Geometrical parameters, potential energy curves (PECs), vibrational energy levels, spectroscopic constants, ionization potentials, and electron affinities of the ground electronic state for all these species are obtained. The information with respect to molecular characteristics of the SFn (n = −1, 0, +1) systems derived in this work will help to extend our knowledge and to guide further experimental or theoretical researches.
Sulfur monofluoride radical is an important species in the fields of atmospheric chemistry and semiconductor industry. The radical is one of the major products of chemical decomposition of SF6, and it takes part in the quenching process in high-voltage power systems.[1] The interest in the SF radical is also attributed to its generation in the plasma discharges employed for etching processes.[2] To better understand the role the radical plays, detailed information with respect to its fundamental properties is required.
The first experimental investigation of SF radical was performed by Carrington et al. with the gas-phase electron paramagnetic resonance technique in 1969.[3] Analyses of the observed spectra yielded equilibrium bond length and effective rotational constant in the ground 2Π3/2 state. The 2Π–2Π transitions were detected by Lonardo and Trombetti in the wavelength region of 4000–3300 Å.[4] Quantitative analyses according to the spectra derived comparable rotational constant with that reported by Carrington et al.[3] Spectrum attributed to SF was also recorded by Hildenbrand with the mass spectroscopy.[5] Based on their analyses, the dissociation energy D0 was obtained. Value of D0 was also determined by Kiang and Zare from the time-of-flight chemiluminescence detection.[6] Improved molecular constants were derived with the microwave spectrometer[7–9] as well as the infrared diode laser spectrometer.[10] The modified RKR method was used by Reddy et al.[11] to determine the vibrational energy levels and dissociation energy of the SF molecule. Theoretically, the methodologies of the HF,[12] CNDO/2,[13] CI,[14] CEPA,[14] DFT,[15–17] G2,[18–20] G3,[20] CCSD(T),[21–24] MRCI,[24–28] and R-matrix[29] were performed for this radical on its geometrical, thermodynamic, and electron collision properties. In these studies, computation results of the equilibrium bond length derived from the CCSD(T) and the MRCI methods with the basis set superior than aug-cc-pVQZ, 1.5983 Å,[24] 1.605 Å,[26] 1.5991 Å,[27] and 1.6040 Å,[28] were closer to experimental results than other approaches, such as MP2, DFT, and CEPA.
In contrast to extensive research on SF radical, much less effort has been made for its singly charged ions, SF+ and SF−. With the aid of the mass spectrometry, Hildenbrand[5] and Fisher et al.[30] provided us the experimental determination of D0 for SF and SF+ with values of 3.51(5) eV and 3.56(5) eV, respectively. To our knowledge, the only experimental datum of SF− for which one can compare was reported by Polak et al.[31] According to the spectrum recorded by a photoelectron spectrometer, Re and ωe of the anion were determined to be 1.717(15) Å and 635(15) cm−1. For both molecular ions, several theoretical investigations were also carried out with HF,[12] MP4,[32] G2,[18–20] G3,[20] DFT,[15–17,33] CCSD(T),[17,21,23] and MRCI[24] methodologies. Among these computations, geometrical parameters obtained on the CCSD(T)/AVQZ,[21] CCSD(T)/cc-pVQZ,[23] and MRCI+Q/AV5Z[24] levels are believed to be trustable than other predictions owing to the high-level methods and relative large basis sets.
In the present work, we have performed high-level computations on the SFn (n = −1, 0, +1) systems to derive their geometrical parameters, spectroscopic constants, PECs, and vibrational energy levels for the ground electronic states. Our aim is to provide accurate information, which are prerequisites for us to understand specific bonding natures, chemical activity, characteristics of photochemistry and thermochemistry, as well as spectroscopic features and reaction dynamics, for systems of interest.
The multi-reference characters of the neutral and ionic SF systems are assessed by utilizing the method of the T1 diagnostics, and the features are found to be quite significant according to our evaluations. Therefore, our computations for these systems are performed at the multi-reference configuration interaction (MRCI) level,[34] based on the complete active space SCF (CASSCF) wavefunctions,[35] with Davidson correction. In order to take into account the electron correlation effect sufficiently, except electrons of F 1s and S 1s are not optimized, all other electrons, including F 2s22p5 and S 2s22p63s23p4, are used in our correlation energy calculations. All energy calculations in this work have been performed with the MOLPRO package.[36] The augmented correlation-consistent basis sets aug-cc-pVXZ (X = Q, 5, 6) of Dunning and co-workers are utilized. The largest basis sets AV6Z are (17s, 11p, 6d, 5f, 4g, 3h, 2i) primitive Gaussian functions contracted to [8s, 7p, 6d, 5f, 4g, 3h, 2i] for F atom, and (22s, 15p, 6d, 5f, 4g, 3h, 2i) to [9s, 8p, 6d, 5f, 4g, 3h, 2i] for S atom. The core–valence (CV) correlation is carried out with the CV basis set aug-cc-pCV5Z, and the relativistic correction is taken into account with the aug-cc-pV5Z-DK basis set coupled with the third-order Douglas-Kroll hamiltonian (DKH3) approximation.[37,38] It should be pointed out that the two correction calculations are applied across the entire PEC.
The corrected total energies are then extrapolated to the CBS limit with the following functions:[39,40]
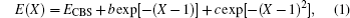

The Murrell–Sorbie (MS) potential energy function[44] is used to represent potential interactions of the present diatomic systems. It is given by

The LEVEL 8.0 program package[45] is used to determine the vibrational energy levels as well as the rotational and centrifugal distortion constants for each level according to our equilibrium geometrical parameters and PECs by numerically solving the one-dimensional Schrödinger equation of nuclear motion.
The PECs of all species are calculated over the internuclear separation range from 0.09 nm to 1.0 nm at intervals of 0.005 nm, which declines to 0.002 nm in the vicinity of the energy minima on each PEC. By fitting energy points to the 15-parameter MS function, PECs of the three systems are obtained and illustrated in Fig.
 | Fig. 1. PECs of the ground states of SF, SF+ and SF−. |
| Table 1. Equilibrium geometrical parameters of SF (X2Π), SF+(X3Σ−) and SF−(X1Σ+). . |
Our final estimation of Re for SF radical, 0.15935 nm, deviates just ∼ 0.17% from the experimental result of 0.1596244(22) nm.[10] Comparing our result with previous theoretical predictions, it is found that our datum agrees with those values quite well, especially with recent CCSD(T)[21–24] and MRCI[24–28] computations. As all other theoretical studies have been done either within small- to moderate-size basis sets or without sufficient consideration of electron correlation effect, our high-level computations are anticipated to yield more trustable predictions.
As for the ionic SF systems, our estimates for Re of SF+ and SF− is 0.14965 nm and 0.17119 nm, respectively. Although several spectroscopic detections were performed, only one study[31] presented experimental determination of Re for SF− and no report of Re was found for SF+ to the best of our knowledge. For SF−, our Re agrees with that reported by Polak et al.,[31] 0.1717(15) nm, quite reasonable. The lack of data made it impossible for us to perform detailed comparisons and further discussion. Nevertheless, from another point of view, this also makes our theoretical result a useful source of information for geometrical parameters of both SF+ and SF−, which should be of interest to experimentalists.
The total energies at the equilibrium internuclear distance of each species are summarized in Table
For further confirmation of the dissociation channel of the ground electronic states of the present systems, we have performed MRCI computations on the ground and several low-lying excited states of both ionic systems. However, as this work focuses on the ground states, no discussion associates with excited states will be presented here. Based on our computation, the dissociation channels of the ground state for the two ionic species are
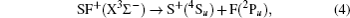

According to our MRCI/CBS calculation, our D0 is 3.627 eV, 3.711 eV, and 3.629 eV for SF, SF+ and SF−, respectively, as listed in Table
Inspection of Table

For all three species, their D0 values are quantitatively similar. The relative large D0 indicates that the neutral FS and its singly charged ions are almost equally strong bounded, and this character of electronic stability ensures the feasibility of performing experimental detections, especially for ionic species, which is a favorable message for spectroscopists. However, to accurate determine D0 of all these species, it is necessary to consider contributions due to nonadiabatic coupling or atomic fine structure and hyperfine structure from successful experimental investigations.[48–51]
Table

| Table 2. Fitting parameters of the MS function and force constants of SF, SF+ and SF− in their ground electronic states. . |
Spectroscopic constants of selected isotopes, 32S19F, 34S19F, 32S19F+, 34S19F+, 32S19F−, and 34S19F−, of the present systems are listed in Table
| Table 3. Spectroscopic parameters of SF, SF+ and SF− (in cm−1). . |
The microwave investigation carried out by Morino and Yamada[9] in 2001 provided accurate spectroscopic constants of SF with Be = 0.5529450(2) cm−1 and centrifugal distortion constant Drot = 9.7484(2) × 10−7 cm−1. It is found that our predictions of both Be and Drot are in good accord with these experimental results with slight deviations of ∼ 0.1%. Our constants of ωe, αe, and ωeχe are also in excellent agreement with those obtained by the infrared study.[10] For example, experimental value of ωe was determined to be 837.6418(13) cm−1. The difference between this result and our estimate is just several wavenumbers. To sum up, our computation results of the neutral SF radical can be considered as a trustworthy source of information. Moreover, this provides evidence of the reliability of our data for both SF+ and SF−.
The vibrational energy levels of the ground electronic states for above-mentioned isotopes are obtained by solving the one-dimensional Schrödinger equation of nuclear motion (with the rotational quantum number J = 0) with the LEVEL program. The rotational constants and high-order centrifugal distortion constants are derived. Table
| Table 4. Vibrational energy levels, rotational constants and centrifugal distortion constants for SF with J = 0 (in cm−1). . |
According to the infrared data,[10] Reddy et al. constructed the PEC of the ground state of SF by using a modified RKR method[11] and derived the vibrational energy levels, which provide us an opportunity to evaluate the reliability of our results. In Table
| Table 5. Vibrational energy levels, rotational constants and centrifugal distortion constants for SF+ and SF− with J = 0 (in cm−1). . |
By using the MRCI method, coupled with the AV6Z basis set, the adiabatic ionization potential (AIP), vertical ionization potential (VIP), adiabatic electron affinity (AEA) and vertical electron affinity (VEA) of the SF radical, as well as the vertical detachment energy (VDE) of the corresponding negative molecular ion, have been determined. Estimations from this work are shown in Table
| Table 6. Ionization potentials and electron affinities of SF and vertical detachment energies of SF− (in eV). . |
We have aimed at theoretical investigations on the SF radical and its singly charged cation and anion, SF+ and SF−. The geometrical structures, potential energy curves, spectroscopic parameters, vibrational energy levels, ionization potentials and electron affinities of the ground electronic state for the three systems have been studied. Computations are performed on the MRCI/aug-cc-pVXZ (X = Q, 5, 6) levels of theory coupled with the technique of CBS extrapolation and consideration of the core–valence correlation and the relativistic effect across the entire PEC. Excellent agreement on geometrical parameters and spectroscopic constants between previous experimental results and our theoretical ones demonstrates the reliability of our data. The information extracted from this work is anticipated to extend our understanding on characteristics of diatomic SF systems, including singly charged molecular ions, and to guide and assist laboratorial detections of these species, which are difficult to exam owing to their nature of high activity.
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 | |
| 22 | |
| 23 | |
| 24 | |
| 25 | |
| 26 | |
| 27 | |
| 28 | |
| 29 | |
| 30 | |
| 31 | |
| 32 | |
| 33 | |
| 34 | |
| 35 | |
| 36 | |
| 37 | |
| 38 | |
| 39 | |
| 40 | |
| 41 | |
| 42 | |
| 43 | |
| 44 | |
| 45 | |
| 46 | |
| 47 | |
| 48 | |
| 49 | |
| 50 | |
| 51 |