Molecular dynamics simulation of Cu
Luo Xianwen1, †,  , Wang Meng1, Hu Bitao2
, Wang Meng1, Hu Bitao2
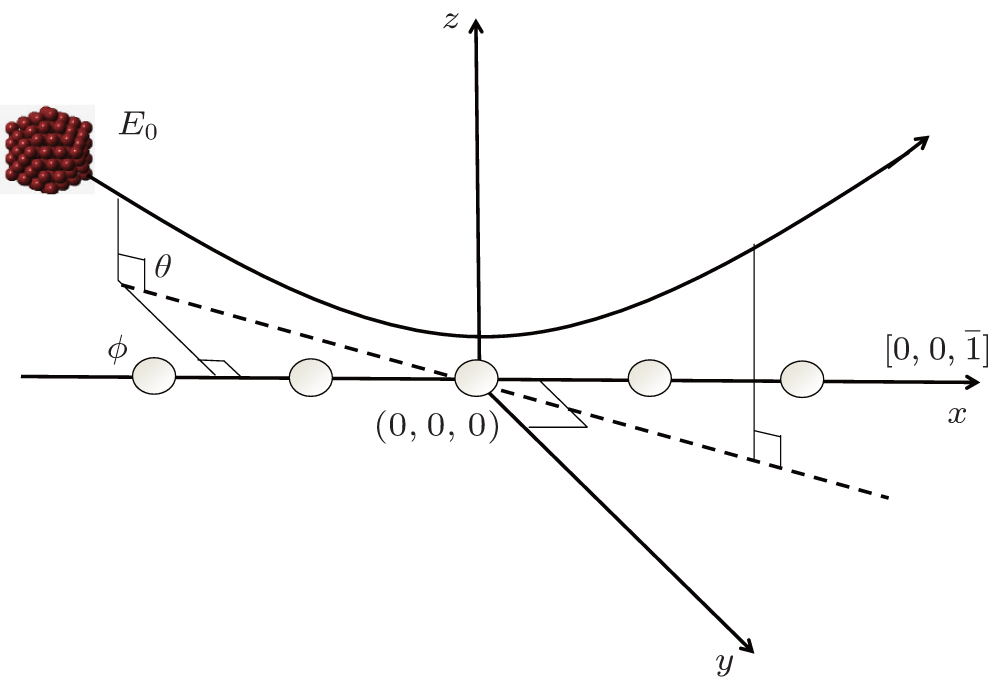
, Wang Meng1, Hu Bitao2 Sketch of the coordinate system.
Molecular dynamics simulation of Cu |
|
Luo Xianwen1, †,
, Wang Meng1, Hu Bitao2 |
Sketch of the coordinate system. |
| |