1. IntroductionRecently there has been considerable interest in developing the solid-state lighting (SSL) device, which is driven primarily by the demands for energy-saving and the concerns of global warming and climate change.[1] As one of the most rapidly evolving branches of SSL technologies, phosphor-converted white light-emitting diodes (pc-white-LEDs) are gradually replacing conventional lighting sources.[2–6] At the present time, the pc-white-LEDs are fabricated by forming a phosphor layer on an output surface of a near ultraviolet (UV) or blue emitting semiconductor chip. The eventual performance of a pc-white-LEDs device strongly depends on the luminescence properties of the phosphor used.[6] Among the various phosphors, the red-emitting phosphors are the most urgent ones to be developed because they can help to convert the cold blue of many current LEDs into the warm white that is preferred for general lighting. The current red-emitting phosphor materials used for pc-white-LEDs include Y2O2S:Eu3+, Eu2+ activated sulfides (e.g., CaS:Eu2+), and Eu2+ or Ce3+-doped (oxy)nitrides (e.g., CaAlSiN3:Eu2+ and CaSiN2:Ce3+).[7–10] Unfortunately, the fluorescent efficiency of Y2O2S:Eu3+ is about eight times lower than those of ZnS:Cu+, Al3+ green, and BaMgAl10O17:Eu2+ blue-emitting phosphors,[7–10] and its lifetime is inadequate under extended UV irradiation. Eu2+-activated sulfide-based red-emitting phosphor is undesirable because of its low chemical stability, and releasing toxic sulfide gas.[7–10] In addition, sulfide-based phosphor shows luminescence saturation with increasing applied current when incorporated into pc-white-LEDs device.[7–10] As for (oxy)nitride-based red-emitting phosphors, high firing temperatures and high nitrogen pressures are required for their synthesis,[11–13] which results in higher production cost. Especially, the 4fn−15d1 → 4fn transitions of Eu2+ and Ce3+ strongly depend on the local crystal field of the surrounding ions in the lattice, which leads to emission band broadening (the emission line widths from these activators are typically 30 nm–80 nm).[7] The full width at half maximum (FWHM) value of the emission band should be as small as possible in order to achieve high luminous output.[7,8] Hence, it is necessary to seek alternative red-emitting phosphor with high absorption in the blue spectral region, small FWHM value, satisfactory chemical stability, and environment friendly properties.
Barium tungstate/-molybdate (BaMO4, M = Mo, W) is a well-known host lattice for rare-earth ions-doped luminescent materials due to their high physical–chemical stabilities and stable crystal structures.[14–29] A good combination of rare-earth ions as activator and barium tungstate/-molybdate host provides us with an excellent luminescent composition emitting a variety of colors. Therefore, BaMoO4 phosphors doped with photo-active lanthanide ions including Eu3+, Sm3+, Tb3+, and Pr3+, have been recently developed.[14–29] Among them, Pr3+-activated BaMoO4 phosphor has received much more attention, because it can be excited by blue light and provides narrow band deep-red emission, resulting in potential application in pc-white-LEDs.[22–29]
Our previous work reported the considerable enhancement of fluorescence from BaMoO4:Pr3+ deep-red-emitting phosphor obtained by the solid state method via incorporation of superfluous alkali metal ions.[23] Despite the above achievement, improvement is still needed to optimize optical properties for potential practical applications. Currently, the forming of a solid solution between two isostructural components is becoming another highly effective strategy for improving luminescence and tuning emission of phosphors.[30–36] However, to the best of our knowledge, optimizing the optical properties for Pr3+-activated barium tungsto-molybdate through the solid solution strategy has not been reported to date. Furthermore, composition-dependent luminescent properties of Pr3+-activated barium tungsto-molybdate have been neglected. Here in this paper, we employ a facile molten-salt method to prepare BaMO4:Pr3+ (M = Mo, W) microcrystals, in which molten NaNO3 acts as a reaction medium at high temperature.[37] Apart from a reaction medium, NaNO3 also acts as a charge compensator. The effects of the substitution of [WO4] for [MoO4] on the structure, morphology, and luminescent properties of BaMoO4:Pr3+ are investigated in detail. Enhanced red emission was observed by the substitution of [WO4] for [MoO4].
2. Experiment2.1. SynthesisThe starting materials including sodium molybdate dihydrate (Na2MoO4·2H2O, 99.9%), sodium tungstate dihydrate (Na2WO4·2H2O, 99.5%), barium chloride dihyrate (BaCl2·2H2O, 99.5%), praseodymium chloride heptahydrate (PrCl3·7H2O, 99.99%), disodium ethylenediamine tetraacetate (Na2H2L, 99.0%), and sodium nitrate (NaNO3, 99.0%) were purchased from Sinopharm Chemical Reagent Co. Ltd, Shanghai, China, and used directly without further purification. Appropriate amounts of Na2MoO4·2H2O, Na2WO4·2H2O, and BaCl2·2H2O were dissolved in distilled water to form aqueous solutions with 0.10-M concentration, respectively. 0.01-M PrCl3 aqueous solution was obtained by dissolving the corresponding PrCl3·7H2O into distilled water.
Pr3+-activated barium tungsto-molybdate phosphors with nominal composition of Ba0.99(Mo1−zWz)O4:0.01Pr3+ (0 ≤ z ≤ 1, in steps of 0.20) were prepared through the combination of the molten-salt process and co-precipitation reaction. Take the synthesis of Ba0.99MoO4:0.01Pr3+ for example. Firstly, a predetermined amount of Na2H2L was added into the mixed solution of 1.98-mmol Ba2+ and 0.02-mmol Pr3+ to form a clear and homogeneous solution of (Ba,Pr)-EDTA complex. Subsequently, 21.00-mL solution containing 2.10 mmol of Na2MoO4·2H2O was introduced dropwise to the above solution under continuous stirring for 10 min. The reaction system was kept still for 2 h at room temperature. Afterward, the pH value of the mixture was adjusted to about 11.0 through the addition of 1%-NaOH solution. Then NaNO3 with a molar ratio of Ba2+:NaNO3 = 1:12 was added into the mixture. The mixture was vigorously stirred for about 30 min to ensure that all reagents were dispersed homogeneously. The as-obtained sample was dried by evaporating water and then put into an alumina crucible with a cover followed by calcination at 500 °C for 6 h in a muffle furnace. After cooling, the solidified melt was thoroughly washed with distilled water at room temperature. Finally, the resulting product was filtered and dried at 120 °C. Ba0.99(Mo1−zWz)O4:0.01Pr3+ microcrystal phosphors were prepared by a similar process.
2.2. CharacterizationPowder x-ray diffraction (XRD) pattern was obtained on a Japan Rigaku D-max 2500 diffractometer, using Ni-filtered Cu Kα radiation (λ = 1.5406 Å). The experimental lattice parameters of Ba(Mo1−zWz)O4:0.01Pr3+ microcrystals were calculated using the least square refinements from the MDI JADE ver. 9.0 software (Materials Data Inc., Livermore CA). Raman spectra were taken with a confocal micro-Raman spectrometer (Super Labram II System, Dilor, France), and the excitation source was a He–Ne laser (632.8 nm). The morphologies of the samples were characterized on a Hitachi S4800 field-emission scanning electron microscope (SEM). Photoluminescence (PL) excitation and emission spectra of powders were recorded using an Edinburgh FLS920 phosphorimeter. For luminescence comparison, the quantity of the phosphor sample was normalized, with the measurement conditions (i.e., the packing density of samples, width of slit of the excitation and emission monochromators, the PMT detector voltage and sensitivity) kept consistent from sample to sample in the measurements. Photoluminescence absolute quantum yield (QY) was determined by employing an integrating sphere (150-mm diameter, BaSO4 coating) from the Edinburgh FLS920 phosphorimeter. All the measurements were performed at room temperature.
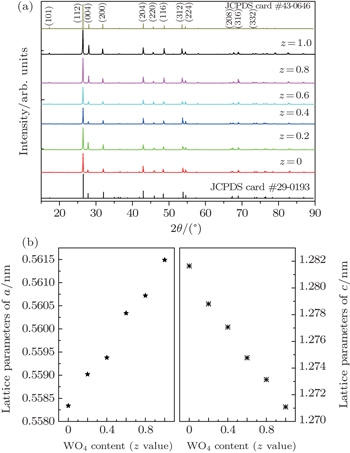
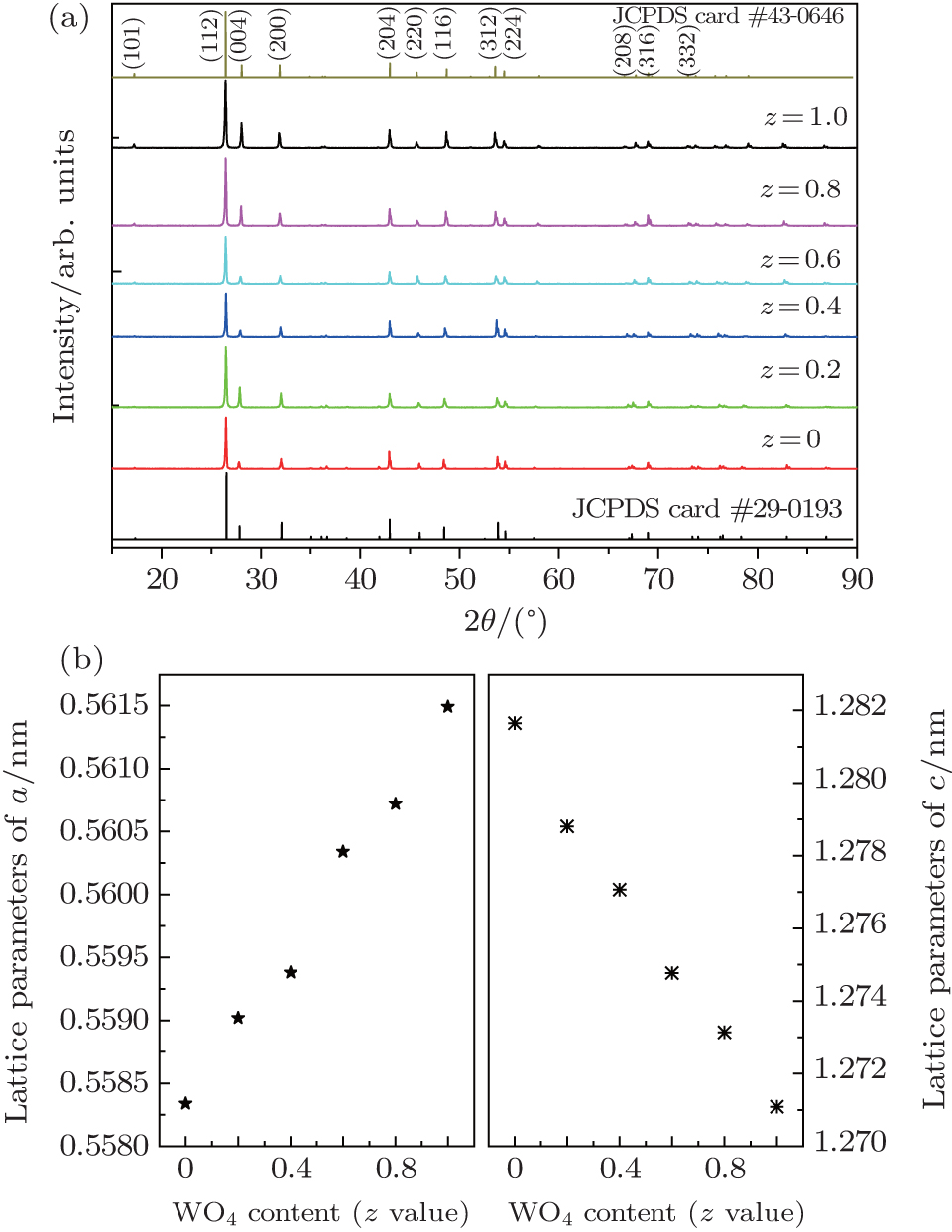
3. Results and discussion3.1. Formation of Ba(Mo1−zWz)O4:Pr3+ solid solution microcrystalsXRD patterns of Ba(Mo1−zWz)O4:0.01Pr3+ (z = 0 ∼ 1, in step of 0.2) series microcrystal powders are shown in Fig. 1(a). For comparison, the standard data for BaMoO4 (JCPDS card No. 29-0193) and BaWO4 (JCPDS card No. 43-0646) are also presented in the figure. The two parent compounds (i.e., BaMoO4 and BaWO4) are isostructural and belong to space group I41/a(88) with a scheelite structure. All the diffraction peaks match well with the standard data for tetragonal scheelite-type Ba(Mo,W)O4 with good crystalline nature. No traces of extra peaks from impurity phases are observed. With increasing the [WO4] content (z value), the (112) diffraction peak shifts obviously toward a smaller angle, while the diffraction peaks become slightly broadened, indicating that the crystal size is gradually reduced. The experimental lattice parameters of Ba(Mo1−zWz)O4:0.01Pr3+ microcrystals are given in Fig. 1(b). A marginal increase in the lattice parameter a value and decrease in the c value are observed. The results reveal that the tungsto-molybdate solid solution is formed upon changing the relative content of [MoO4] and [WO4].
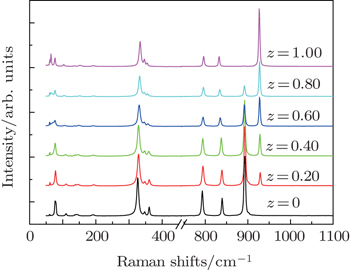
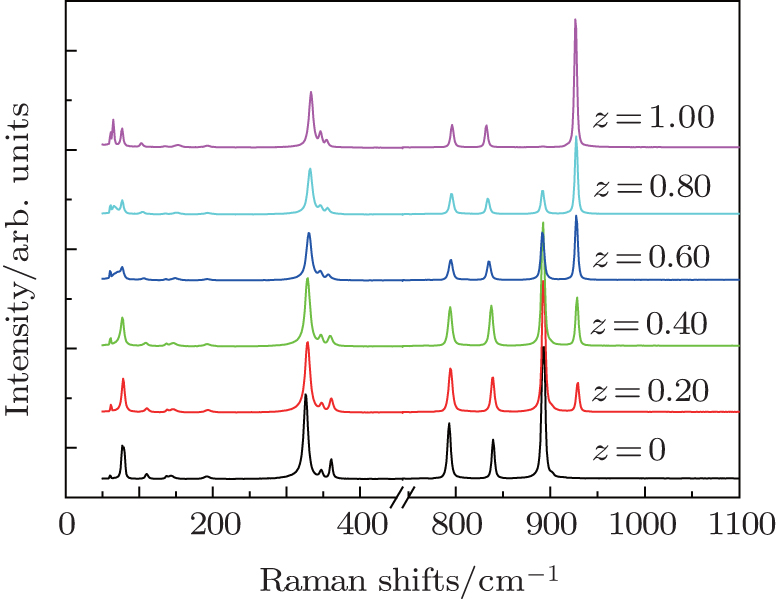
As indicated above, the as-obtained BaMO4:Pr3+ (M = Mo, W) series microcrystals maintain a sheelite crystal structure with tetragonal symmetry 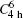 at room temperature. Its primitive cell includes two formula units. Owing to weak coupling between the [MO4] (M = Mo, W) group and Ba2+ cations, the vibrational modes in Raman spectra of Ba(Mo,W)O4 sheelite crystals can be divided into two groups, internal and external.[38–41] The internal vibrons correspond to the oscillations inside the [WO4] or [MoO4] group with an immovable mass center. The external or lattice phonons relate to the motion of the Ba2+ ions and the rigid molecular unit. There are 26 different vibration modes in the sheelite primitive cell: Γ = 3Ag + 5Au + 5Bg + 3Bu + 5Eg + 5Eu, among which only the even vibrations (Ag, Bg, and Eg) are Raman-active.[38–41] Raman spectra of Ba(Mo1−zWz)O4:Pr3+ samples are shown in Fig. 2. Factor group assignments of the Raman-active modes are summarized in Table 1.[38–41]
at room temperature. Its primitive cell includes two formula units. Owing to weak coupling between the [MO4] (M = Mo, W) group and Ba2+ cations, the vibrational modes in Raman spectra of Ba(Mo,W)O4 sheelite crystals can be divided into two groups, internal and external.[38–41] The internal vibrons correspond to the oscillations inside the [WO4] or [MoO4] group with an immovable mass center. The external or lattice phonons relate to the motion of the Ba2+ ions and the rigid molecular unit. There are 26 different vibration modes in the sheelite primitive cell: Γ = 3Ag + 5Au + 5Bg + 3Bu + 5Eg + 5Eu, among which only the even vibrations (Ag, Bg, and Eg) are Raman-active.[38–41] Raman spectra of Ba(Mo1−zWz)O4:Pr3+ samples are shown in Fig. 2. Factor group assignments of the Raman-active modes are summarized in Table 1.[38–41]
Table 1.
Table 1.
 Table 1. Frequencies and assignments of Raman-active modes for Ba(Mo1−zWz)O4: Pr3+ microcrystals. .
| Lattice mode |
Ba(Mo1−zWz)O4: 0.01Pr3+ |
Assignments |
symmetry 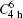 |
z = 0 |
z = 0.20 |
z = 0.40 |
z = 0.60 |
z = 0.80 |
z = 1.0 |
| Ag |
|
929 |
928 |
927 |
927 |
926 |
ν1 (A1) |
|
892 |
892 |
892 |
891 |
891 |
|
|
| Bg |
839 |
839 |
837 |
834 |
833 |
832 |
ν3 (F2) |
| Eg |
793 |
794 |
794 |
795 |
796 |
796 |
ν3 (F2) |
| Eg |
360 |
361 |
359 |
356 |
356 |
354 |
ν4 (F2) |
| Bg |
347 |
348 |
346 |
345 |
345 |
346 |
|
| Bg |
327 |
328 |
328 |
330 |
332 |
333 |
ν2 (E) |
| Eg |
192 |
193 |
192 |
192 |
192 |
192 |
νf.r. (F1) |
| Ag |
143 |
145 |
146 |
148 |
150 |
154 |
|
| Bg |
136 |
137 |
137 |
136 |
135 |
134 |
νext. |
| Eg |
110 |
109 |
108 |
105 |
104 |
102 |
|
| Bg |
80 |
|
|
71 |
65 |
64 |
WO4, MoO4, |
| Eg |
76 |
78 |
77 |
77 |
77 |
76 |
and Ba2+ motions |
f.r.: free rotation; ext: external modes
| Table 1. Frequencies and assignments of Raman-active modes for Ba(Mo1−zWz)O4: Pr3+ microcrystals. . |
The well-resolved sharp peaks of Raman spectra indicate that all the samples are highly crystallized, which is in agreement with above XRD results. According to group theory analysis, the Ag peak is due to the stretch vibration of the oxygen ion of the [MO4] (M = Mo, W) group. With substitution of W6+ for Mo6+ in Ba(Mo1−zWz)O4:Pr3+ series phosphors, the new peak at a higher wavenumber (926 cm−1) from the [WO4] group is present and enhanced, while the 892-cm−1 peak for [MoO4] is weakened. These results further confirm that the solid solutions are formed between end members BaMoO4 and BaWO4.
3.2. Morphology evolution of Ba(Mo1−zWz)O4:Pr3+ solid solution microcrystalsFigure 3 shows the SEM images of Ba(Mo1−zWz)O4:Pr3+ microparticles with various z values. The surfaces of the microcrystals are smooth without any small particles attached on them. As shown in Fig. 3(a), for the BaMoO4:Pr3+ sample, most of the particles are of a truncated octahedral shape with 2.3 μm ∼ 4.2 μm in arris length and 4.5 μm ∼ 7.6 μm hemline length. With the z value increasing to 0.20 (Fig. 3(b)), uniform octahedrons are almost the exclusive products. When the content of [WO4] further reaches 0.40, a large scale of microcrystals with a size of about 2.0 μm is obtained as displayed in Fig. 3(c). With the further increasing of the [WO4] content, the crystals lose octahedral shapes and their sizes decrease. In this work, after melting the reaction mixtures, complete dissolution of reactants occurs in all cases, forming the transparent solutions. Consequently, the morphologies of the crystals are due to the growth from homogeneous solutions and unrelated to the shapes of precursor particles. Since neither surfactants nor templates are used, the growth habit of BaWO4 crystal accounts for the evolution of the morphology with increasing [WO4] content.[42,43]
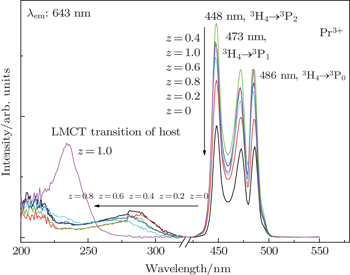
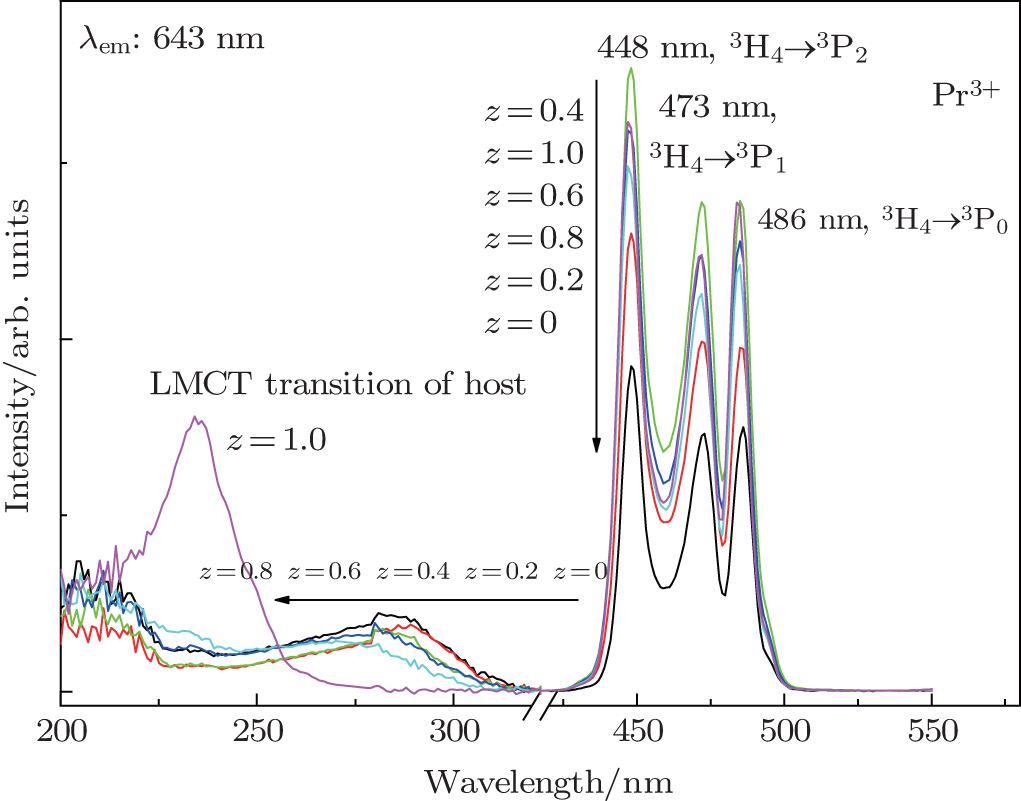
3.3. Photoluminescent characteristics of Ba(Mo1−zWz)O4: Pr3+ phosphorsExcitation spectra of Ba(Mo1−zWz)O4:0.01Pr3+ microcrystals (see Fig. 4) are composed of a broad band and three strong peaks in the 435 nm–500 nm range. The presence of strong peaks indicates the high crystallinity of the as-obtained micropaticles, which is very beneficial to achieving stronger luminescence. The broad band can be attributed to the ligand to metal charge-transfer (LMCT) transitions of [MoO4] and [WO4] groups.[21,23,30] With the increase of [WO4] concentration (i.e., z value), the LMCT band of [MoO4] gradually shifts towards short wavelengths, which indicates that the correlations among [MoO4] groups exist although they are separated by BaO6 groups. With the substitution of W6+ for Mo6+, the average distance between [MoO4] groups increases and the electron delocalization among [MoO4] groups decreases, thus the blue shift of the LMCT band is observed. On the other hand, the Raman shift value (i.e., wave number, ν) of a vibration mode has the following relation with the bonding strength (k) and the reduced atom mass (m):
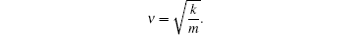
The reduced atom mass for the [WO4] group is larger than that for [MoO4], but the Ag peak related to WO4 shifts towards a higher wavenumber (see Fig. 2), which implies that the W–O bond is stronger than Mo–O. Accordingly, it is deducted that the stronger bond of W–O than that of Mo–O leads to Eg in WO4 being larger than that in MoO4, which accounts for the blue shift of the LMCT excitation band accompanied with increasing of the [WO4] content.
As shown in Fig. 4, the sharp excitation peaks at 448, 473, and 486 nm are due to 3H4 → 3P2, 3H4 → 3P1, and 3H4 → 3P0 transitions of Pr3+ ions, respectively. With the increase of the [WO4] concentration (z value), the excitation peaks of 4f–4f transitions of Pr3+ ions are enhanced. When z reaches 0.4, the corresponding microparticle exhibits the largest intensity.
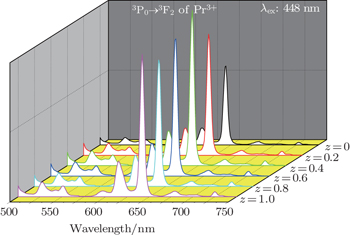
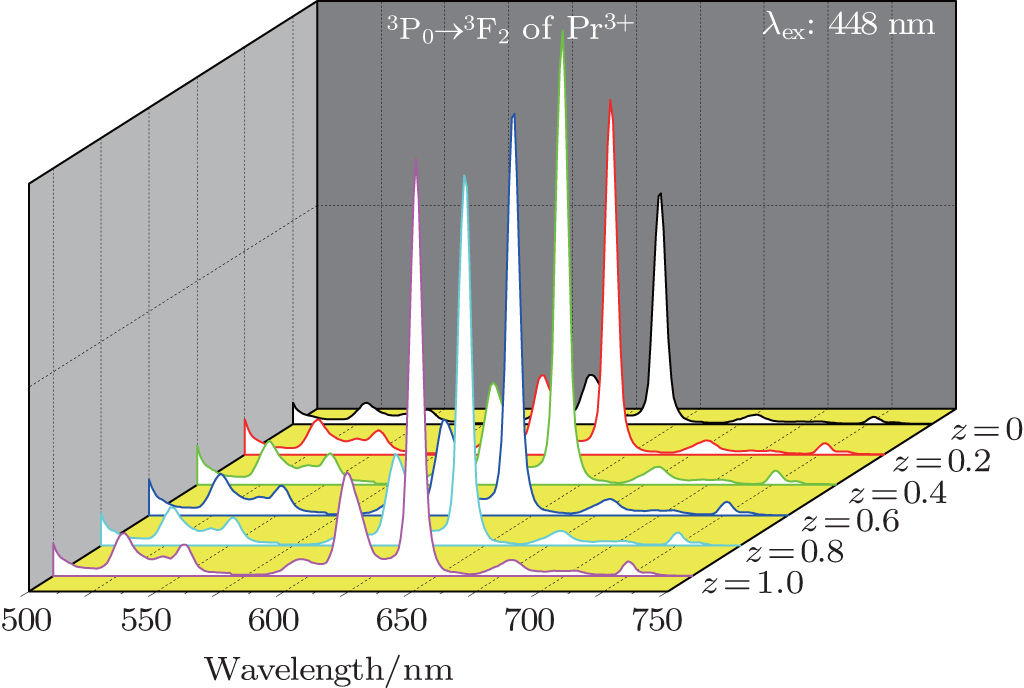
Figure 5 shows the emission spectra of Ba(Mo1−zWz)O4:0.01Pr3+ microparticles under 448-nm excitation. The spectra contain two main peaks at 643 nm and 615 nm from Pr3+ ions. However, no emission corresponding to tungstate or molybdate is observed. The presence of an absorption band from the tungstate or molybdate group in the excitation spectra (see Fig. 4), indicated by monitoring the Pr3+ emission (643 nm), suggests that the energy absorbed by the tungstate or molybdate group is transferred to Pr3+ levels nonradiatively. This has been known as a host-sensitized energy transfer process.[15,17,18] However, the intensity of Pr3+ emission is weaker upon CT band excitation than that under Pr3+ direct excitation, which reveals that the energy from the host lattice to activator Pr3+ is incomplete. As indicated in Fig. 5, the substitution of [WO4] for [MoO4] leads to the enhancement of luminescence intensity. The deep-red emission intensity of Pr3+3P0 → 3F2 transition reaches a maximum when the relative ratio of [MoO4] to [WO4] is 3:2. Ba(Mo0.60W0.40)O4:0.01Pr3+ exhibits the strongest deep-red emission in this series of samples. The possible reasons for this enhancement are as follows. With the substitution of W6+ for Mo6+, the phase structure remains unchanged, while the size and surface of microparticles become more and more uniform and smooth, respectively. Hence the improved luminescence is exhibited.
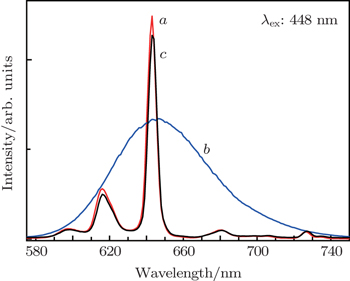
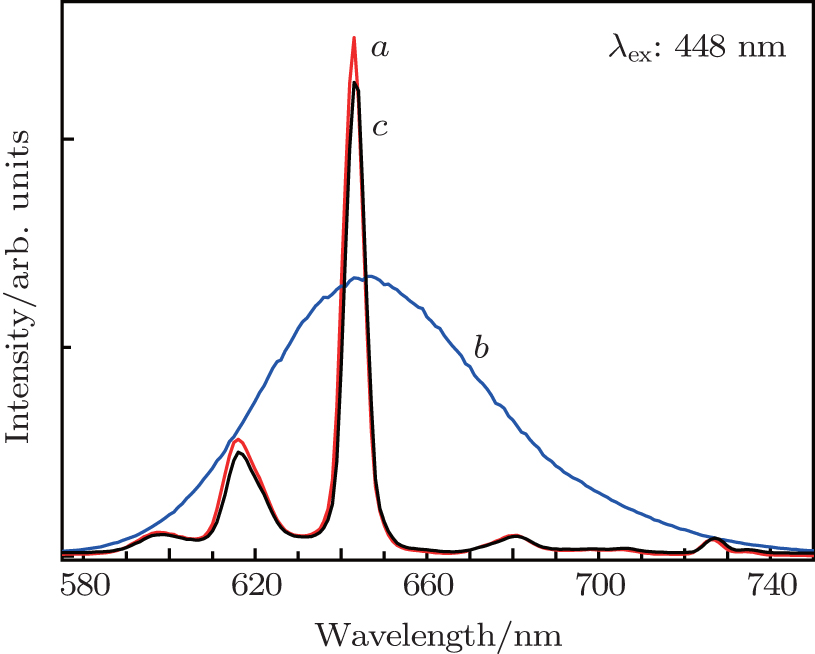
3.4. Evaluation of performance for composition-optimized solid solution deep-red-emitting phosphorTo evaluate the performance of the above solid solution phosphor, the photoluminescence properties of as-obtained Ba(Mo0.60W0.40)O4:0.01Pr3+ are compared with those of a commercial deep-red-emitting phosphor CaS:Eu2+ and BaMoO4:0.01Pr3+, 0.10Na+ phosphor (abbreviated as BMP) with the same doping level of Pr3+, recently reported by our group.[21] As shown in Fig. 6(b), CaS:Eu2+ exhibits a broadband emission with a peak at around 643 nm, which is due to the eg → t2g transition of Eu2+ ions.[44] Comparing curve a with curve b of Fig. 6, under the same excitation wavelength, 643-nm emission intensity of Ba(Mo0.60W0.40)O4:0.01Pr3+ is about 1.86 times higher than that of commercial CaS:Eu2+. However, the integral luminescence intensity of the former is about 36% lower than that of the latter. As revealed in curves a and c of Fig. 6, its intensity is 1.09 time higher than that of BMP. In addition, the absolute QY of (Mo0.60W0.40)O4:0.01Pr3+ microparticles is determined to be 57% bigger than that of BMP (50%), but slightly lower than that of CaS:Eu2+ (62%).
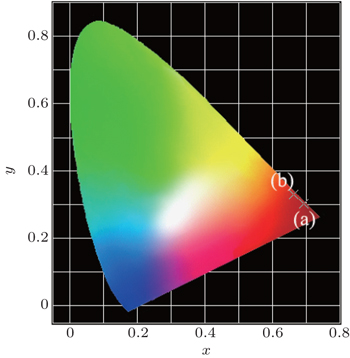
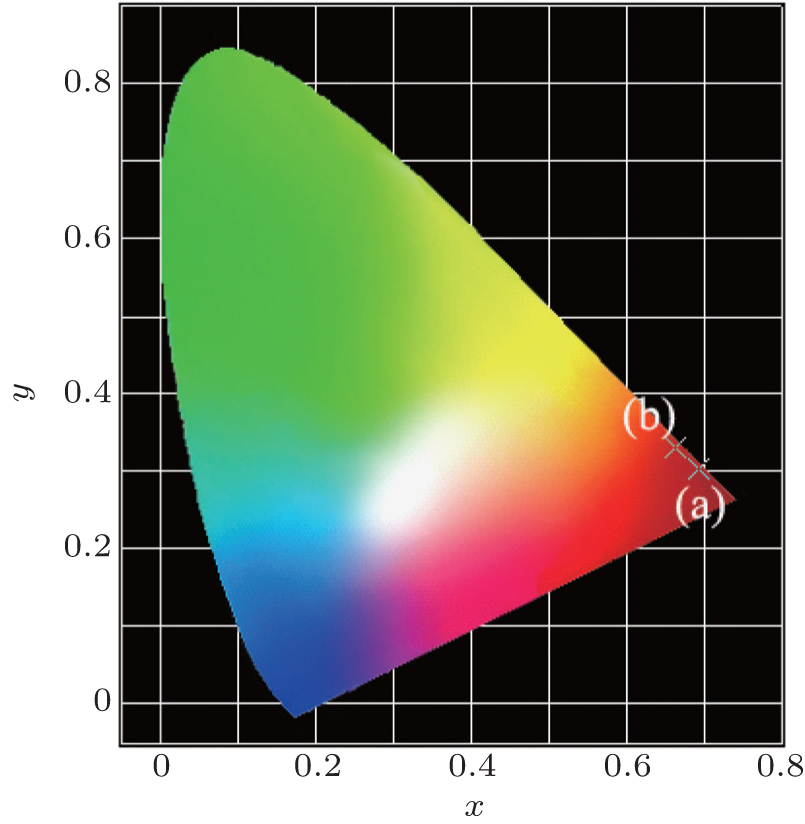
The Commission Internationale de L’Eclairage (CIE) chromaticity coordinates of Ba(Mo0.60W0.40)O4:0.01Pr3+ and CaS:Eu2+ samples are calculated to be (x = 0.693, y = 0.302) and (x = 0.662, y = 0.331), respectively, located in the deep-red light region (Fig. 7). Especially, the chromaticity point of Ba(Mo0.60W0.40)O4:0.01Pr3+ is close to the edge of the CIE diagram.
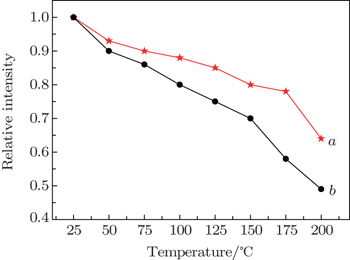
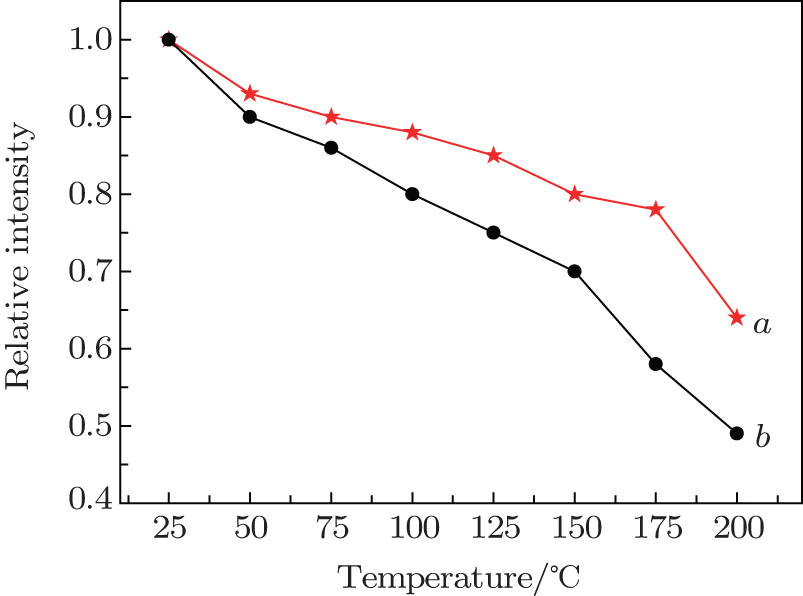
The temperature stability is one of the most important technological parameters for phosphors used in pc-white-LEDs.[4,6,7,13] The temperature-dependent PL properties of as-prepared Ba(Mo0.60W0.40)O4:0.01Pr3+ and CaS:Eu2+ phosphors are investigated in a temperature range from 25 °C to 200 °C, and the results are shown in Fig. 8. With increasing temperature, the deep-red emission intensity gradually decreases due to the thermal quenching.[4,6,7,13] At 150 °C, luminescent intensities of Ba(Mo0.60W0.40)O4:0.01Pr3+ and CaS:Eu2+ are still about 80% (this value is higher than that previously reported by Yang et al.[19]) and 70% of those measured at room temperature, respectively. Up to 200 °C, the relative emission intensities of Ba(Mo0.60W0.40)O4:0.01Pr3+ and CaS:Eu2+ phosphors maintain 64% and 49% of their initial values, respectively. These results confirm that Ba(Mo0.60W0.40)O4:0.01Pr3+ has excellent thermal stability and its temperature stability is desirable for SSL applications. In addition, this phosphor is free of sulfur pollution, which has intrinsic properties of commercial deep-red-emitting phosphor CaS:Eu2+.
4. ConclusionsIn this study, BaMO4:Pr3+ (M = Mo, W) solid solution microparticles with high crystallinity and excellent luminescent properties, are prepared through a simple molten-salt process at 500 °C. The structural, morphology, and optical properties of as-obtained microcrystal phosphors are investigated each as a function of the [WO4] content. The microparticles remain truncated octahedral morphologies with uniformity in size with increasing [WO4] content. The substitution of [WO4] for [MoO4] of BaMoO4 host can significantly improve its luminescence, which may be attributed to the shape evolution and improvement of particle size distribution. The integral emission intensity and absolute QY of composition-optimized Ba(Mo0.60W0.40)O4:0.01Pr3+ are lower than those of a commercial deep-red-emitting phosphor CaS:Eu2+. However, Ba(Mo0.60W0.40)O4:0.01Pr3+ has excellent thermal stability and is free of sulfur pollution. The combination of high phase purity, well-defined facets, highly uniform morphologies, exceptional chemical and thermal stabilities, and stronger emission intensity makes the resulting solid solution sample a potential deep-red-emitting phosphor for the pc-white-LEDs application.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 , Ye Zhao-Lian1, 2, Guan Ming-Yun1, Lian Ning1, Sun Jian-Hua1]
, Ye Zhao-Lian1, 2, Guan Ming-Yun1, Lian Ning1, Sun Jian-Hua1]