First-principles calculations of structural and electronic properties of Tlx Ga1−xAs alloys
[Akyüz G. Bilgeç1, †,  , Tunali A. Y.2, Gulebaglan S. E.3, Yurdasan N. B.2]
, Tunali A. Y.2, Gulebaglan S. E.3, Yurdasan N. B.2]
, Tunali A. Y.2, Gulebaglan S. E.3, Yurdasan N. B.2]
|
|


† Corresponding author. E-mail:
The zincblende ternary alloys TlxGa1−xAs (0 < x < 1) are studied by numerical analysis based on the plane wave pseudopotential method within the density functional theory and the local density approximation. To model the alloys, 16-atom supercells with the 2 × 2 × 2 dimensions are used and the dependency of the lattice parameter, bulk modulus, electronic structure, energy band gap, and optical bowing on the concentration x are analyzed. The results indicate that the ternary TlxGa1−xAs alloys have an average band gap bowing parameter of 4.48 eV for semiconductor alloys and 2.412 eV for semimetals. It is found that the band gap bowing strongly depends on composition and alloying a small Tl content with GaAs produces important modifications in the band structures of the alloys.
Semiconductors which have interesting electronic properties are used as important optoelectronic devices (laser diodes, detectors) and the progresses in the optoelectronic technology requires the development of new materials. III–V type semiconductor materials are relatively easy to synthesize, so these materials have been intensively studied by using theoretical and experimental methods. Especially, thallium-V compounds, which have been offered as important materials for long-wavelength infrared detectors[1] and heterostructure field-effect transistors,[2] have attracted considerable interest in recent years for electronic applications and optoelectronic devices. The theoretical[1,3,4] and experimental works have provided the opportunity to study the growth, electronic and structural properties of thallium-V compounds. Improvements in the experimental procedures (MBE, MOVPE) have evoked the studies about the growth and electronic properties of thallium-V compounds and these systems have been performed many times in the early studies to investigate their structural stability.[5]
TlxGa1−xAs alloys were able to be synthesized[6] and grown[7,8] in the experimental investigations. Also these alloys have been examined in numerical studies.[3,9] Zhou et al.[9] have calculated the band structure and total energy of their electronic structure, by employing first principles calculations within density functional theory (DFT) and generalized gradient approximation (GGA). In their studies, they have examined 8-atom cubic unit cell TlnGa4−nAs4 and pronounced that the gap bowing of TlxGa1−xAs shows a significant increase with the structural relaxation increasing, which is primarily attributed to the larger atomic size difference between Ga and Tl. Mankefors and Svensson[3] also performed ab-initio calculations of the Ga1−xTlxAs alloys. It has been reported that the lattice constant and bulk modulus each had a positive dependence on the Tl concentration in the ternary system. The aim of the present study is to investigate bowing parameters and the origin of band gap bowing parameter in detail, and to study more thoroughly the electronic structures and bonding properties of these alloys.
It is expected that Tl–V compounds should have small or negative gaps due to the Tl band gap properties. Additionally, the combination of Tl with III–V compounds yields interesting new semiconductor ternary alloys. These new alloys show different characters from semiconductors to semi-metal compounds. Measurements and calculations of semiconductor alloys indicate that the band gap energy of the materials deviates from the behavior given by Vegard’s law.[10] The addition of the second or higher order term is necessary for this deviation. This add-on term is known as the band-gap bowing parameter. This parameter determines the alloy band-gap energy and is a measure of the nonlinearity of the energy gap functional dependence on composition. For instance, we have reported a composition dependent bowing parameter for GaxTl1−xP alloys[11] in our previous study. For the GaxTl1−xP alloys, the band gap increases with increasing the composition of Ga, even the compounds changing from metal to semiconducting phase.
The band gap bowing parameter is a crucial parameter to determine the alloy band-gap energies and it is a measure of the nonlinearity of the energy-gap functional dependence on composition. Experimental techniques (photoluminescence (PL), absorbtion) give different band gap values for the ternary alloys.[12] To better understand the physical origins of the large dispersion and composition-dependent bowing in TlxGa1−xAs alloys, we essentially investigate how the band gap bowing varies as a function of alloy composition. In the present study, we present a systematic and comparative study of the structural and electronic properties of the zincblende TlxGa1−xAs (x = 0.125–0.875) alloys. Firstly, we compute the lattice constants and energy band gaps of the binary TlAs and GaAs compounds for the zincblende phase by using local density approximation (LDA) for exchange-correlation potential. Ternary compounds are constituted by means of a 16-atom supercell, and the composition dependence of lattice constants, density of states, energy band gap and physical origin of the band gap bowing parameter are represented.
In our study, the structural and electronic properties of TlAs and GaAs compounds and TlxGa1−xAs alloys are investigated by using the plane wave pseudopotentials methods within the PWSCF software package[13] which is based on DFT[14,15] and pseudopotentials. Many DFT-based first-principles studies using plane wave ultrasoft pseudopotentials have been also performed in the early studies for different materials.[16,17] Exchange and correlation potentials are described within the LDA. Wave functions are expanded in a plane wave basis to an 80-Ryd cutoff energy. Ga (4s24p1), As (4s24p3), and Tl (5d106s26p1) valance electrons are used, and spin–orbit interactions are ignored throughout the calculations. Accurate Brillouin zone investigations are performed by the Monkhorst-Pack method with a 14 × 14 × 14 special k-point for binary compounds and 12 × 12 × 12 meshes for ternary alloys.
The TlxGa1−xAs alloys with x = 0.125, 0.25, 0.375, 0.5, 0.625, 0.75, and 0.875 are performed by using a 16-atom supercell in zincblende structure which corresponds to a 2 × 2 × 2 conventional cubic cell. In the calculations, firstly, the GaAs supercell is constituted and then Tl atoms are doped into this supercell to a desired concentration. Tl and As atoms are distributed randomly in the unit cells of TlxGa1−xAs alloys. The different random distribution models of Tl and As atoms in the unit cell for a fixed value of x do not exhibit any change for structural nor electronic quantities of TlxGa1−xAs alloys. All alloy configurations studied are relaxed by geometry optimization at their optimum volumes. All forces on the atoms are converged to less than 2.0 mRy/a.u., in the geometrical optimization.
We first examine the structural properties of the GaAs and TlAs zincblende phases which constitute the ternary alloys. The ground state structure of TlAs is determined as a zincblende phase in the early investigations.[5] Lattice constants, bulk modulus, and gap energies of binary compounds are computed by fitting the calculated total energies with respect to different volumes to the Vinet’s equation[18] of state (EOS). Results are given in Table Values of lattice parameter a (Å), band gap energy (Eg), and bulk modulus (B0) for the TlAs and GaAs in zincblende structure phase at equilibrium volume. Available pseudopotential data are also shown for comparison.
a/Å
Eg/eV
B0/GPa
TlAs (present work)
6.169
−0.104
51.535
TlAs[1]
6.18
−2.12
…
TlAs[3]
6.27
−1.86
…
TlAs[9]
6.088
…
55.18
TlAs[19]
6.170
…
50.2
TlAs[23]
6.052
0.000
…
GaAs (present work)
5.548
1.354
76.254
GaAs[9]
5.596
…
76.58
GaAs[12]
5.653
1.42
…
GaAs[23]
5.164
1.0475
…
GaAs[20] (Exp.)
5.653
1.519
…
The band gap structures give the direct band gap Γ–Γ values of −0.104 eV and 1.354 eV for TlAs and GaAs, respectively. Band gap results are in general agreement with other theoretical calculations (Table Band structures of zincblende GaAs and TlAs along the high symmetry directions in the Brillouin zone with LDA.
The lattice parameters, band gap energies and bulk modulus which we obtained for different concentrations of Tl in the TlxGa1−xAs ternary alloys are listed in Table Calculated values of lattice parameter a (Å), band gap energy (Eg), and bulk modulus (B0) for TlxGa1−xAs ternary alloys in zincblende structure at equilibrium volume.
x
a/Å
Eg/eV
B0/GPa
0.00 GaAs
5.548
1.354
76.254
0.125
5.625
0.761
71.718
0.25
5.703
0.017
67.538
0.375
5.783
0.00
63.571
0.5
5.863
−0.026
60.179
0.625
5.939
−0.044
57.551
0.75
6.015
−0.145
55.237
0.875
6.092
−0.115
52.908
1 (TlAs)
6.169
−0.104
51.535
To proceed further with the analysis of the amount of deviation from the Vegard’s law, we compare the results in Fig. Composition dependences of the calculated lattice constant (a) and bulk modulus (b) of the ternary alloys compared with Vegard’s law predictions.
The lattice parameter data can be approximated by the following form:
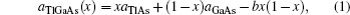
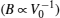
To determine the electronic properties of the system, we calculate the band structures of binary and ternary alloys along the high symmetry direction in the first Brillouin zone (BZ) for the calculated equilibrium lattice constant values. For some specific values of x, we plot the electronic band structures of the ternary alloys along the high symmetry points in BZ in Fig.
Figure Electronic band structures along the high symmetry direction in the Brillouin zone for TlxGa1−xAs alloys with (a) x = 0.25, (b) x = 0.5, and (c) x = 0.75. Valance band maximum is the zero energy level. Total and partial DOS components calculated for TlxGa1−xAs alloys in equilibrium volume with (a) x = 0.25, (b) x = 0.5, and (c) x = 0.75.
When the dos graphics are analyzed, it is generally observed that p-electrons of the Tl, Ga, and As atoms dominate in the valance and conduction bands. However, s orbitals of Tl and Ga atoms are only effective in the lowest group bands for the ternary alloys and there is a negligible effect of Tl-5d electrons on the studied ternary alloys. It is observed that As-4p states dominate the valance bands in all the samples. Additionally, as expected, the contributions of the thallium electrons to the valance band increase while the gallium electrons contributions decrease with increasing x concentration. In the conduction band, the contributions of Tl-4s and Tl-4p states increase while the contributions of Ga-4p states decrease and As contributions stay nearly stable with increasing Tl concentration. The crucial point which is observed in the band and dos graphs is that the conduction band width narrows with increasing x value and even disappears for large x-values.
The band gap energy of the ternary alloy can be depicted as a function of the band gap energy of the binary compound and determined with the following formula:[23]
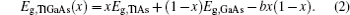
The overall band gap bowing parameters of TlxGa1−xAs alloys are also calculated from the equations of Bernard and Zunger[24] for 0 < x < 1. In this approach, the overall bowing coefficient at a given average composition x measures the change in the band gap according to the formal reaction
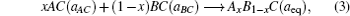
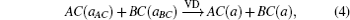
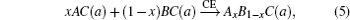
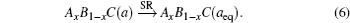
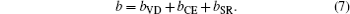
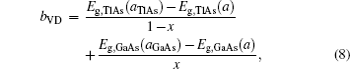
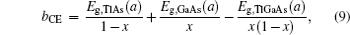
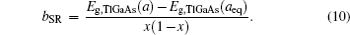
Composition dependences of calculated band gap bowing parameter b (a) and three contributions bVD, bCE, and bSR (b).
Contributions of volume deformation, charge exchange, structural relaxation and formation energies of the ternary alloys are listed. Calculated band-gap bowing parameters (bZunger) of TlxGa1−xAs ternary alloys are compared with that obtained by quadratic fit (bQuad). All bowing parameters are in eV and formation energies are in Ry/f.u.
| x | 0.125 | 0.25 | 0.375 | 0.5 | 0.625 | 0.75 | 0.875 |
|---|---|---|---|---|---|---|---|
| bVD | 2.761 | 2.757 | 2.690 | 2.500 | 1.889 | 1.453 | 1.203 |
| bCE | 1.031 | 2.419 | 0.755 | 0.104 | 0.188 | 0.704 | 0.563 |
| bSR | −0.036 | 0.011 | 0 | 0 | 0 | 0.005 | 0 |
| bZunger | 3.756 | 5.187 | 3.445 | 2.604 | 2.077 | 2.162 | 1.766 |
| bQuad | 3.771 | 5.189 | 3.449 | 2.604 | 2.081 | 2.154 | 1.771 |
| Eform | −12.812 | −18.098 | −24.754 | −28.674 | −33.950 | −39.225 | −44.502 |
As seen from Table
Additionally, in order to study the stabilities of TlxGa1−xAs alloys, we calculate the formation energies of all studied alloys (Table
We also investigate the bonding properties of the ternary alloys to complete the basic background of the study. We analyze the charge density distributions to obtain an insight into the electronic natures of the alloys. The electronic charge density contours in the (110) crystallographic plane are shown for some special concentration values (x = 0.25, 0.5, and 0.75) in Fig. Electronic charge density distribution contours calculated in the (110) crystallographic planes of TlxGa1−xAs compounds with (a) x = 0.25, (b) x = 0.5, and (c) x = 0.75. Electronic charge density distribution contour calculated in the (101) crystallographic planes of Tl0.5Ga0.5As compound.
Figure Electronic localization function (ELF) contour calculated for the (101) crystallographic plane of Tl0.5Ga0.5As alloy.
We investigate the structural and electronic properties of TlAs, GaAs, and TlxGa1−xAs alloys by utilizing numerical analysis based on first-principles calculations. We first determine that the structural and electronic properties of the binary compounds are in good agreement with available theoretical and experimental results. To investigate the structural and electronic properties of zincblende TlxGa1−xAs alloys, we calculate the band-structure, lattice parameter, electronic density of states, charge density graphs and gap bowing parameter. It is found that the relaxed lattice constants of the ternary alloys are very compatible with the calculations from Vegard’s law and a small downward bowing parameter equal to 0.006 Å is obtained. However, we obtain a large upward bulk modulus bowing parameter equal to 14.139 GPa because of the distinctive difference in bulk modulus between the GaAs and TlAs. The band gap energies are obtained for various alloy compositions through the electronic band-structure calculations, and it is observed that TlxGa1−xAs alloys have a semiconductor character for x = 0.125 and x = 0.25 concentration values and semi-metalic character for other concentration values. We find that the ternary TlxGa1−xAs alloys each have an average band gap bowing parameter of 4.48 eV for semiconductor ones and 2.412 eV for semimetals.
Substitution of thallium for GaAs changes the materials from the semiconductor phase to the semimetalic phase and leads to a large and composition dependent band-gap bowing parameter. According to this important effect, our study can be useful for enhancing the performances of the optoelectronic devices and for further studies.
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 | |
| 22 | |
| 23 | |
| 24 |