{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
First-principle investigation on the thermodynamics of X2N2O (X = C, Si, Ge) compounds
[Xiong Qing-Yun1, Shen Qi-Xia2, Li Rui-Zi1, Shen Jiang1, Tian Fu-Yang1, †,  ]
]
]
|
|


† Corresponding author. E-mail:
Project supported by the Fundamental Research Funds for the Central Universities, China (Grant No. FRF-TP-14-029Al).
The structures under different pressures, elastic properties, electronic structures and lattice vibrations of the X2N2O (X = C, Si, Ge) compounds are investigated by using the first-principle method. Based on the phonon density of state, the thermodynamic properties of the present compounds are studied under different pressures and at different temperatures. The structural parameters including the bond lengths and bond angles are in agreement with available experimental measurements and theoretical calculations. We employ the elastic theory to calculate the nine independent elastic constants (Ci j) and the derived elastic moduli (B, G, E, v). Results indicate that these X2N2O (X = C, Si, Ge) compounds are mechanically stable and show the brittle behaviors. The electronic properties of the present compounds are analyzed by using the band structure and density of states. The phonon dispersion calculations imply that the present compounds are dynamically stable. Based on the quasi-harmonic approximation, the calculations of the specific heat indicate that the temperature in a range of 0 K–1500 K and pressure in a range of 0 GPa–40 GPa have a large effect on the thermal quantities of Ge2N2O, compared with on those of the C2N2O and Si2N2O compounds.
Silicon oxynitride (Si2N2O) and germanium oxynitride (Ge2N2O) are widely used as the high-temperature structural materials and viewed as the promising functional materials.[1–3] Much attention has been paid to the superior physical and chemical properties, such as good thermal shock resistance, high mechanical strength and excellent oxidation resistance.[4–7] The hypothetical carbon oxynitride C2N2O has the same crystal structure as the Si2N2O and Ge2N2O compounds.[8,9] It was theoretically predicted that the mechanical properties of C2N2O are superior to Si2N2O and Ge2N2O.[3,10]
Most of theoretical and experimental reports focus on structural and mechanical properties of X2N2O (X = C, Si, Ge). For example, the electronic structures and chemical bondings of these compounds were studied by the orthogonalized linear combination of atomic orbitals (OLCAO) method[11] and the full-potential linear Muffin-tin orbitals (LMTO) method.[10] The results showed that C2N2O has stronger covalent bonding than Si2N2O and Ge2N2O. Ding[3] and Ding et al.[12] studied the hardness values of the X2N2O (X = C, Si, Ge) and found that the C2N2O may be the potential superhard material, whereas the hardness of Ge2N2O is the smallest in these three compounds. On the experimental side, extensive studies focused on the preparation and improving mechanical properties of Si2N2O ceramic.[4,7,13] Wu and Li[1,14] indicated the relationship between purity and Vickers hardness of Si2N2O with the final sintering temperature. More recently they successfully fabricated a nano-grained Si2N2O ceramic without second phases. Srinivasa and Cariz,[5] and Srinivasa et al.[15] determined the lattice parameters, bond properties of Si2N2O and Ge2N2O at pressure P = 2.3 and 2.54 GPa by the time-of-flight neutron diffraction. Haines and Santoro[16] found the complete amorphization of Si2N2O occurs above P = 41 GPa by the x-ray power diffraction and the infrared absorption. Although the electronic properties and thermodynamic properties of some ceramics were reported,[17–22] as a widely used refractory, however, few experimental and theoretical data about the thermodynamic properties of X2N2O (X = C, Si, Ge) compounds have been reported so far.
In the present work, we systematically investigate the structures under different pressures, elastic properties, and electronic structure of X2N2O (X = C, Si, Ge) using the first-principle calculations. The thermodynamic properties are calculated via quasi-harmonic approximations under different pressures (0 GPa–40 GPa) and at various temperatures (0 K–1500 K). The lattice parameters, average bond lengths and bond angles are presented in detail. The results of elastic constants and lattice vibration show that these compounds are mechanically and dynamically stable and the derived mechanical moduli are further studied. The band structures of these compounds are calculated and the contributions of different orbitals to density of state (DOS) are discussed. The specific heat Cv values are obtained at different temperatures (0 K–1500 K) and under different pressures (0 GPa–40 GPa).
The rest of the paper is organized as follows. In Section 2 we present the theoretical tool and give the important details of the numerical calculations. The results are presented and discussed in Section 3. And finally, we draw some conclusions from the present studies in Section 4.
All first-principle calculations were carried out using Cambridge Series of Total Energy Package (CASTEP) code based on the density functional theory (DFT).[23] The effects of the exchange–correlation energy were described by the generalized gradient approximation (GGA) in the form of Predew–Burke–Ernzerhof (PBE) potential.[24] The optimization was performed until the forces on the atoms are less than 0.01 eV/Å and all the stress components were less than 0.05 GPa. Ultrasoft pseudo potentials were used. The electronic wave functions were expanded in a plane-wave basis set with a cutoff energy of 400 eV, and 8 × 13 × 13 Monkhorst–Pack meshes were employed for the Brillouin-zone k-point samplings. The self-consistent convergence of the total energy was 5.0 × 10−6 eV/atom. The finite displacement method was employed to calculate the phonon of state and the phonon dispersion via the constructed 3 × 3 × 3 supercell.
The stress-strain method was used to evaluate the elastic constants of the crystal structure. Owing to the crystal symmetry, the present compounds can be described by the nine independent elastic stiffness coefficients (C11, C22, C33, C44, C55, C66, C12, C13, and C23). For calculating the elastic constants, we used the volume-conserving strains. The maximum strain amplitude is set to be 0.003. The number of steps is 6 for each strain. The mechanical parameters such as bulk modulus (B), shear modulus (G), Young’s modulus (E), and Poisson’s ratio (v) can be calculated via Voigt–Reuss–Hill (VRH) approximation.[25] For the orthorhombic crystal, the Voigt bound of bulk modulus (BV) and the Voigt bound of shear modulus (GV) in Voigt approximation are calculated directly from elastic constants by
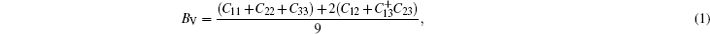
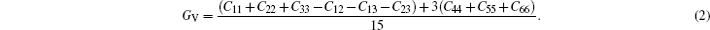
In Reuss approximation, the Reuss bound of bulk modulus (BR) and the Voigt bound of shear modulus (GR) are calculated by
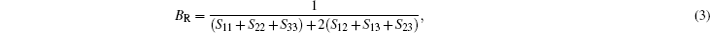
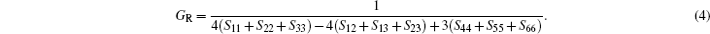
In the formulas above, the Si j are elastic compliance constants, which are the inverse matrix of Ci j. In Voigt–Reuss–Hill method, the bulk moduli B and shear moduli Gare calculated by arithmetic average of Voigt and Reuss bounds as follows:
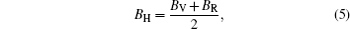
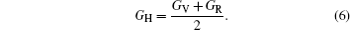
Then E and v can be calculated from the following equations:
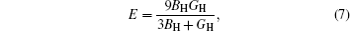
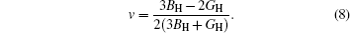
As is well known, the calculations of phonon spectra can be used to compute energy (E), entropy (S), free energy (F), and lattice specific heat (CV) each as a function of temperature via the quasi-harmonic approximation.[26] The temperature dependence of the energy can be expressed as
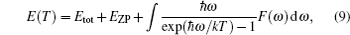
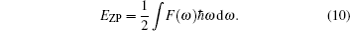
The vibrational contribution to the free energy F(T) is expressed as
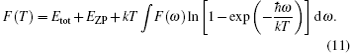
The lattice contribution to the specific heat Cv is written as
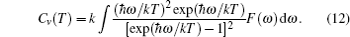
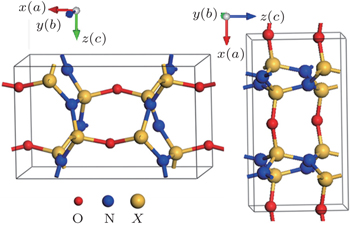
The X2N2O (X = C, Si, Ge) compounds are of the orthorhombic structure (shown in Fig.
 | Fig. 1. Crystal structures of the orthorhombic- X2N2O (X = C, Si, Ge) compounds. |
| Table 1. Comparisons among the calculated values of equilibrium lattice parameters (a, b, c) (Å), average bond length L (Å), and bond angle (°), the experimental data and other theoretical results. For each compound, the data in the first line are from the present calculations, the data in the second line are the results cited from the references, and the data in the third line refer to the experimental results. . |
For X2N2O (X = C, Si, Ge), one oxygen atom and three nitrogen atoms form a tetrahedron structure, and the X element is located in the center of the tetrahedron structure. With the increase of atomic radius of X atom (X = C, Si, Ge), the average X–N and X–O bond lengths increase, while the X–O average bond length is shorter than the X–N ones. Whereas the average bond angles of O–X–N and N–X–N are still 109°. As is well known, the sp3 hybrid in the carbon group results in the tetrahedron structure with a bond angle of 109°. The bond angle of X–O–X in Si2N2O is large, compared those of the C2N2O and Ge2N2O compounds. The N–X–N bond angle is about 120°for the three present compounds.
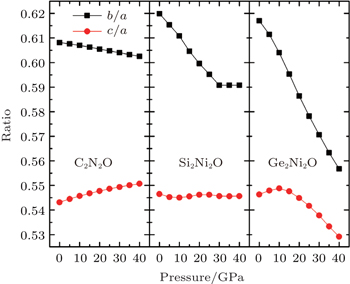
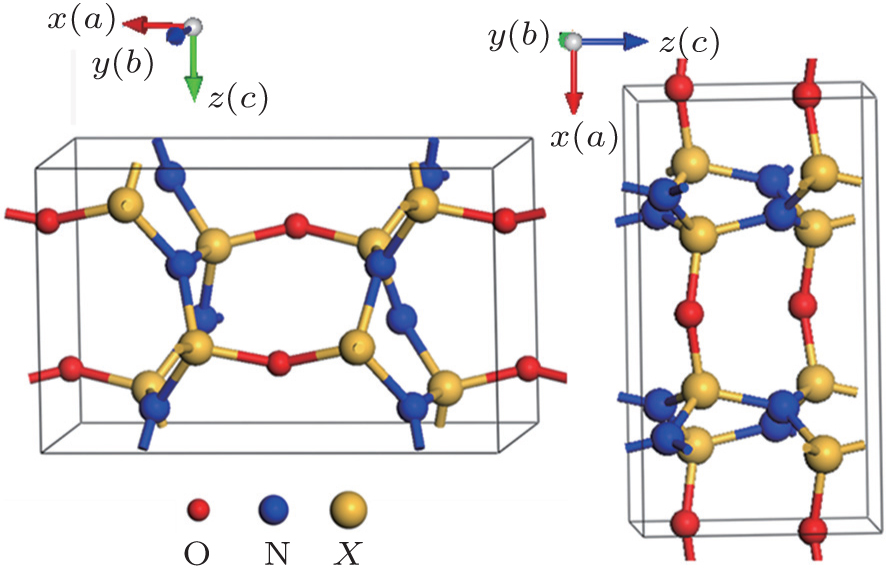
Considering the fact that the lattice parameters (a, b, c) of each compound are not equal to each other, we show in Fig.
Comparative investigations of these structures are helpful to understand the effects of C and Ge on the elastic and thermodynamic properties in these compounds. The equilibrium structures under zero pressure are used for discussing the elastic properties, band structures, phonon information, and thermodynamic properties.
 | Fig. 2. Variations of lattice parameter ratio (b/a and c/a) of crystal structure with pressure for the orthorhombic- X2N2O (X = C, Si, Ge) compounds. |
Elastic constants (Cij) are significant parameters of solid materials, which provide important information about the mechanical stability, anisotropic feature, bonding characteristics, ductility, etc.[31] Listed in Table
The calculated values of shear modulus G, Young’s modulus E, and Poisson’s ratio ν are listed in Table
| Table 2. Calculated elastic constants Cij (GPa) and elastic moduli (GPa) of the orthorhombic-X2N2O (X = C, Si, Ge) compounds. . |
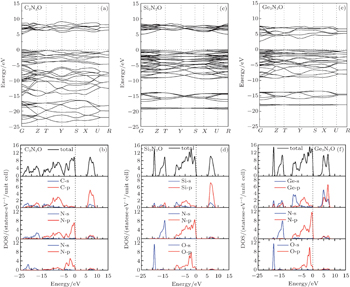
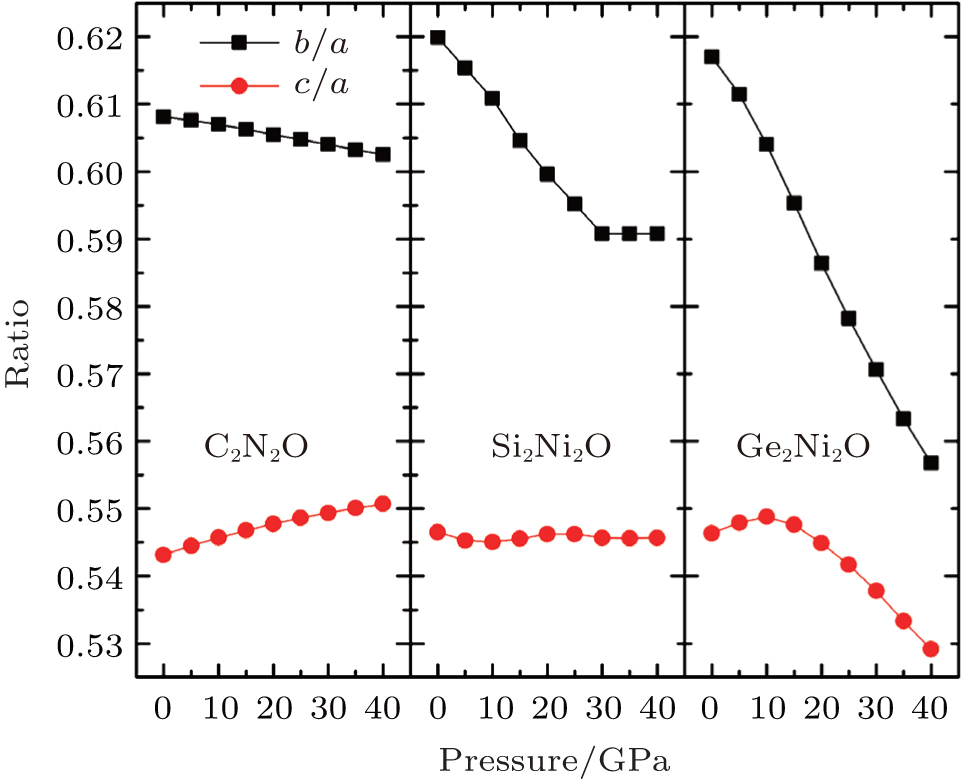
The calculated band structures and densities of state (DOSs) of X2N2O (X = C, Si, Ge) are shown in Fig.
Figures
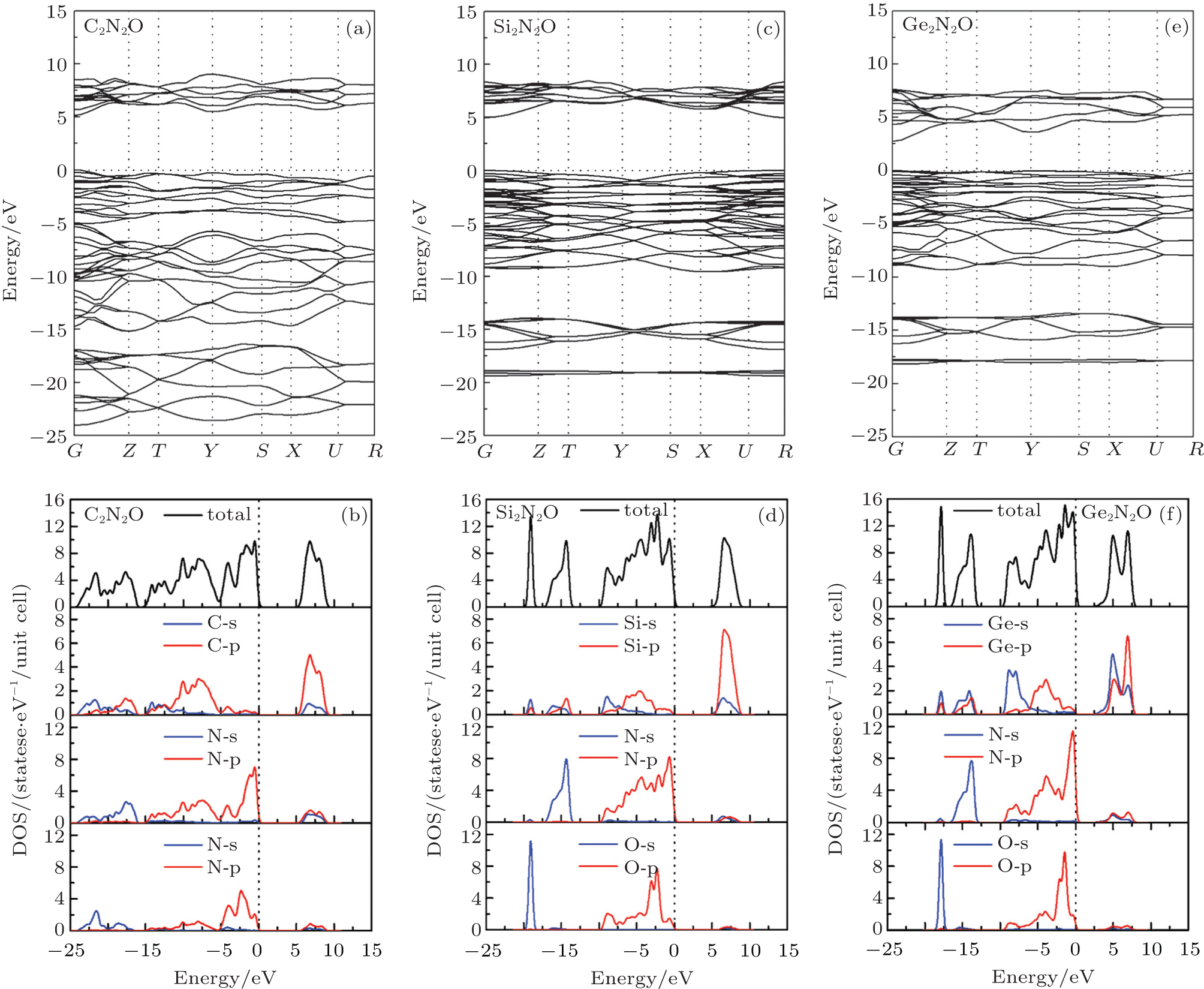 | Fig. 3. Band structures and DOSs for X2N2O (X = C, Si, Ge). |
Figure
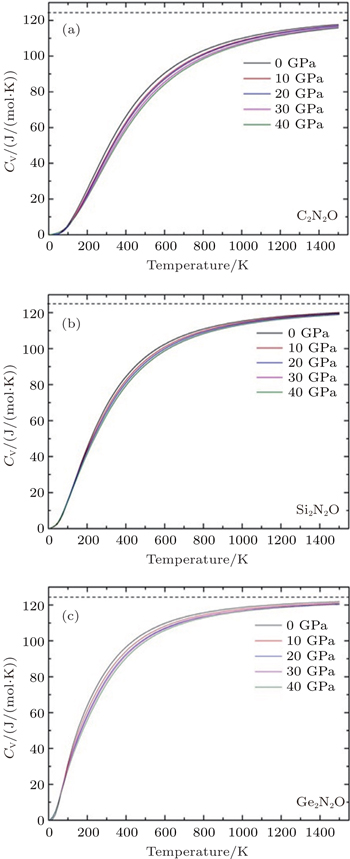
The specific heat is a measure of how well the substance stores the heat. The calculated values of specific heat CV as a function of temperature for all the investigated compounds at different pressures are shown in Fig.
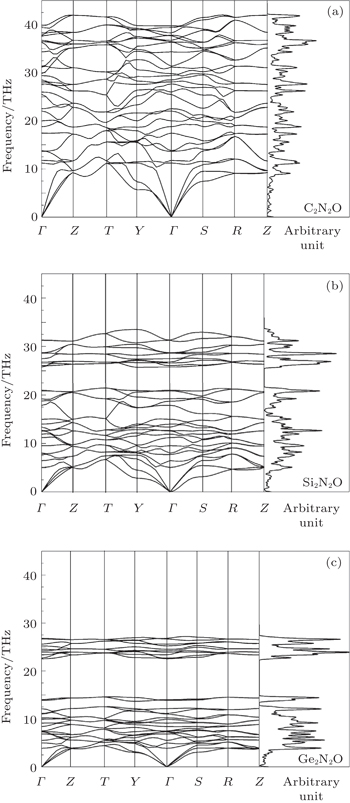
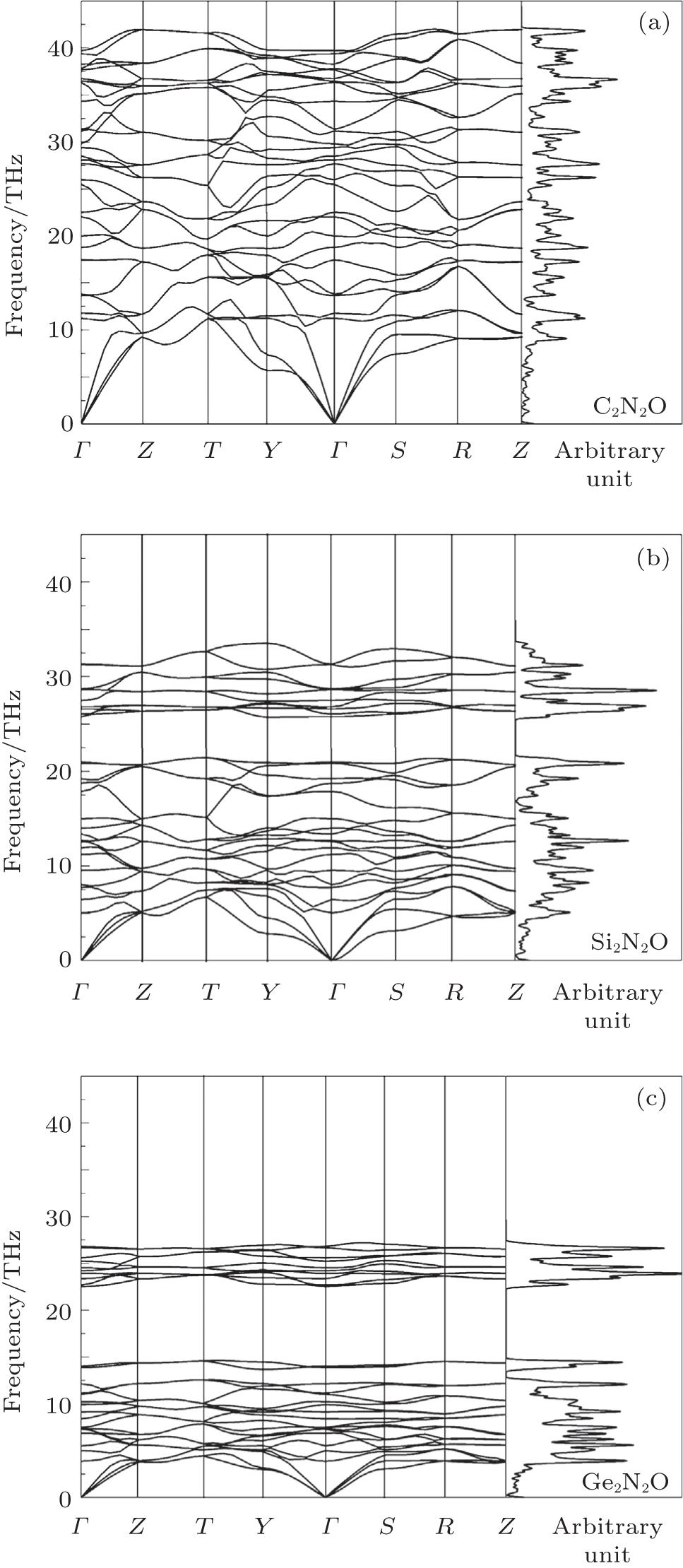 | Fig. 4. Phonon dispersions and phonon DOSs for X2N2O [X = C, Si, Ge, corresponding to panels (a), (b), and (c)]. |
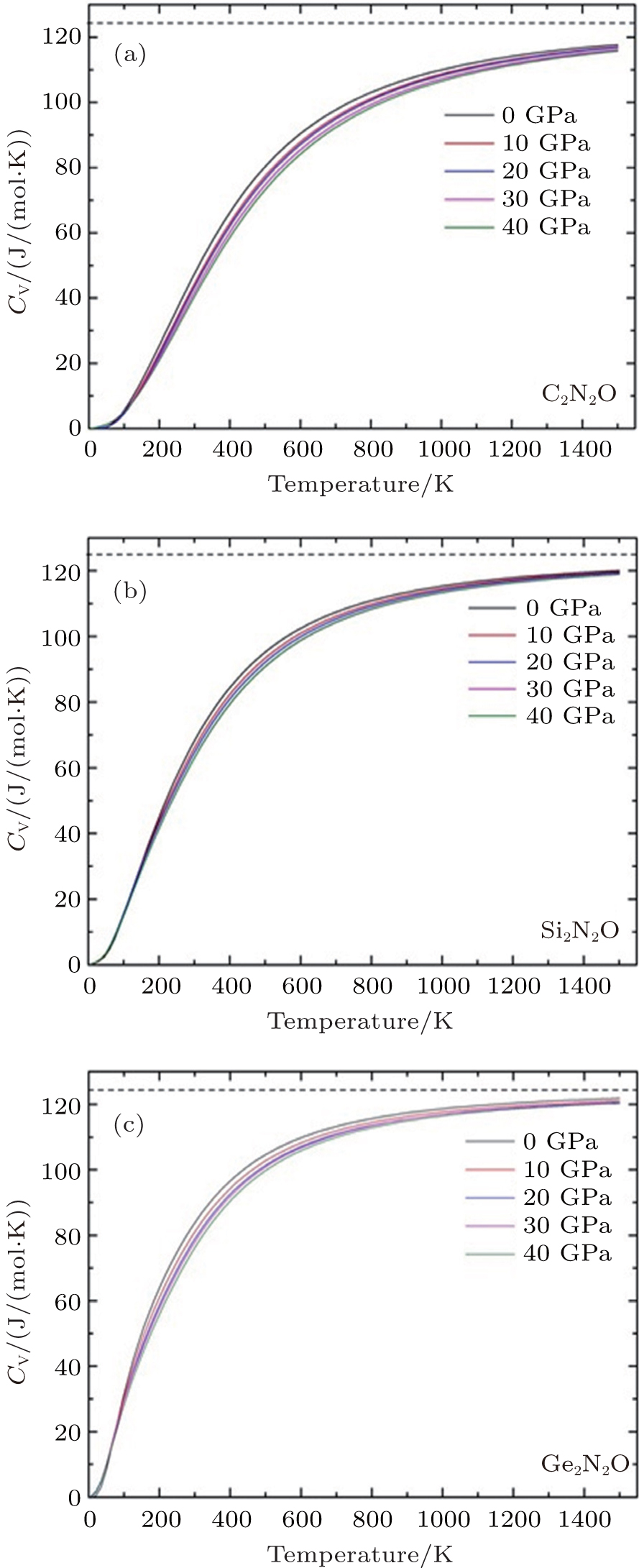 | Fig. 5. Variations of Cv with temperature T at different pressures for X2N2O [X = C, Si, Ge, corresponding to panels (a), (b), and (c)]. |
In this work, the crystal structures, elastic properties, electronic structures and thermodynamic properties of X2N2O (X = C, Si, Ge) are investigated using the first-principle method. The orthorhombic structure is thermodynamically stable indicated by the calculations of the elastic constants and the lattice vibration. The obtained results of the structural parameters, elastic constants (Cij), and aggregate elastic moduli (B, G, E) are in agreement with the corresponding experimental and theoretical results. The lattice parameters each as a function of pressure imply the strong anisotropies of X2N2O. The different values of band gap for X2N2O (X = C, Si, Ge) are mainly attributed to the X-s and p orbitals. The specific heat CV values are calculated at different temperatures and under different pressures via the quasi-harmonic approximation. Compared with the cases of the Si2N2O and C2N2O compounds, the specific heat at constant volume of Ge2N2O remarkably increases with increasing temperature at given pressure before it is close to the Dulong–Petit limit.
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 | |
| 22 | |
| 23 | |
| 24 | |
| 25 | |
| 26 | |
| 27 | |
| 28 | |
| 29 | |
| 30 | |
| 31 | |
| 32 | |
| 33 | |
| 34 |