Zhang Han, Jin Shifeng, Guo Liwei, Shen Shijie, Lin Zhiping, Chen Xiaolong. Bandgap narrowing in the layered oxysulfide semiconductor Ba3Fe2O5Cu2S2: Role of FeO2 layer. Chinese Physics B, 2016, 25(2): 026101
Permissions
Bandgap narrowing in the layered oxysulfide semiconductor Ba3Fe2O5Cu2S2: Role of FeO2 layer
Zhang Han1, Jin Shifeng1, †, , Guo Liwei1, Shen Shijie1, Lin Zhiping1, Chen Xiaolong1, 2
Research and Development Center for Functional Crystals, Beijing National Laboratory for Condensed Matter Physics, Institute of Physics, Chinese Academy of Sciences, Beijing 100190, China
Collaborative Innovation Center of Quantum Matter, Beijing, China Research & Development Center for Functional Crystals, Beijing National Laboratory for Condensed Matter Physics, Institute of Physics, Chinese Academy of Sciences, Beijing 100190, China
Project supported by the National Natural Science Foundation of China (Grant Nos. 51472266, 51202286, and 91422303), the Strategic Priority Research Program (B) of the Chinese Academy of Sciences (Grant No. XDB07020100) and the ICDD.
Abstract
Abstract
A new layered Cu-based oxychalcogenide Ba3Fe2O5Cu2S2 has been synthesized and its magnetic and electronic properties were revealed. Ba3Fe2O5Cu2S2 is built up by alternatively stacking [Cu2S2]2− layers and iron perovskite oxide [(FeO2)(BaO)(FeO2)]2− layers along the c axis that are separated by barium ions with Fe3+ fivefold coordinated by a square-pyramidal arrangement of oxygen. From the bond valence arguments, we inferred that in layered CuCh-based (Ch = S, Se, Te) compounds the +3 cation in perovskite oxide sheet prefers a square pyramidal site, while the lower valence cation prefers the square planar sites. The studies on susceptibility, transport, and optical reflectivity indicate that Ba3Fe2O5Cu2S2 is an antiferromagnetic semiconductor with a Néel temperature of 121 K and an optical bandgap of 1.03 eV. The measurement of heat capacity from 10 K to room temperature shows no anomaly at 121 K. The Debye temperature is determined to be 113 K. Theoretical calculations indicate that the conduction band minimum is predominantly contributed by O 2p and 3d states of Fe ions that antiferromagnetically arranged in FeO2 layers. The Fe 3d states are located at lower energy and result in a narrow bandgap in comparison with that of the isostructural Sr3Sc2O5Cu2S2.
Layered Cu-based oxychalcogenides have received considerable attention due to their promise in areas including transparent conducting materials,[1–4] thermoelectric materials.[5–7] and photocatalysts,[8] etc. The two-dimensional (2D) nature of these compounds, the (CuCh2)2− layer in particular, is the origin of the interesting transport and optical properties. The wide applications of these compounds are mainly determined by their bandgaps which are tunable. As a narrow band gap semiconductor, BiCuSeO has high mobility originating from the Cu 3d/S 3p antibonding states and low thermal conductivity, making it a promising candidate for commercial thermoelectric applications.[5–7] Meanwhile, the isostructural quaternary oxychalcogenides LnCuOS (Ln = La–Nd) are optically transparent in the visible region and are used as p-type transparent conducting semiconductor.[1] Other promising candidates for wide-band-gap semiconductors are those in which perovskite-type oxide layers containing d0 or d10 metals separate the antifluorite-type [Cu2Ch2]2− layers. For example, Sr2ZnO2Cu2S2, Sr3Sc2O5Cu2S2, and their derivatives have been explored in the context of viable transparent p-type conductors, with bandgaps up to 3.1 eV.[2,3] However, when the d10 metals in the oxide layers of Sr2ZnO2Cu2S2 are replaced by magnetic atoms Co or Mn and the S atoms substituted by Se, a bandgap narrowing occurred and the optical bandgaps of Sr2CoO2Cu2Se2 and Sr2MnO2Cu2Se2 were drastically decreased to 0.068 eV and 0.073 eV, respectively.[9] The reason, however, responsible for this effective bandgap narrowing is still unclear.
The bandgap widths in Cu-based oxychalcogenides are generally correlated with the electronegativity of chalcogenide, as well as the basal lattice parameter. In LaOCuCh (Ch = S, Se, Te) system, the bandgaps decrease from 3.1 eV to 2.31 eV with reducing the electronegativity of chalcogenide from S to Te.[4] While for a given chalcogenide ions, a decrease of the basal lattice parameter from 4.067 Å to 3.879 Å in LnOCuSe (Ln = Y and La) also results in slight narrowing of the bandgap (from 2.82 eV to 2.58 eV).[10,11] However, the bandgap narrowing observed in Sr2CoO2Cu2Se2 and Sr2MnO2Cu2Se2 is exceptional, since such a drastic narrowing effect was not observed in other nonmagnetic oxychalcogenides systems. A natural speculation then arises that the 3d electrons in the perovskite-type oxide layers might be more or less responsible for such a bandgap narrowing. Therefore, it is desirable to find a new 3d transitional metal oxysulfide to verify this speculation and study the mechanism of bandgaps narrowing in layered Cu-based oxychalcogenides. Here, we report the synthesis and characterization of a new layered Cu-based oxysulfides, Ba3Fe2O5Cu2S2, which is isostructural to nonmagnetic Sr2Sc3O5Cu2S2 without d electrons. Interestingly, compared with the wide bandgap of 3.1 eV in Sr2Sc3O5Cu2S2, the optical bandgap of Ba3Fe2O5Cu2S2 significantly reduced to 1.03 eV. Our theoretical calculations indicated that Ba3Fe2O5Cu2S2 should be a metal without considering the effect of spins, while it becomes a narrow bandgap mott insulator if considering the spin-polarized AFM magnetic interaction. The conduction band minimum (CBM) of Ba3Fe2O5Cu2S2 is comprised of the Fe 3d/O 2p states and is considerably lowered in compared with the isostructural Sr3Sc2O5Cu2S2, which is responsible for the observed bandgap narrowing.
2. Material and methods
The Ba3Fe2O5Cu2S2 sample was prepared by the reaction of BaO (99.9%), FeO (99.9%), Cu (99.99%), and S (99.999%) powders in stoichiometric ratio. Reagents were mixed together and sealed inside an evacuated quartz tube. The sealed quartz tube was heated slowly to 1123 K and held at this reaction temperature for 24 h before furnace-cooling to room temperature. Powder x-ray diffraction (PXRD) was performed at room temperature using a PANalytical X’pert Pro diffractometer with Cu Kα radiation. Rietveld refinement was performed using the FULLPROF program.[12] DC magnetization M(T) and Resistivity data were measured on a Physical Properties Measurement System (PPMS, Quantum Design) using powders. The diffuse reflectance spectra of the samples were measured on a UV-3600 Plus ultraviolet-visible light-near-infrared (UV-vis-NIR) spectrophotometer over the range 220 nm–2600 nm. Reflectance spectra were converted to absorbance expressed as F(R) using the Kubelka–Munk function. The first principles calculations were performed using the CASTEP program.[13] The generalized gradient approximation (GGA) in the form of the Perdew–Burke–Ernzerhof was chosen to solve the exchange-correlation potentials.[14] The ultrasoft pseudopotential with a plane-wave energy cutoff of 380 eV and a Monkhorst Pack k-point separation of 0.04 Å−1 in the reciprocal space were used for all the calculations.[15] We employed the “LDA + U” (LDA: local density approximation) correction with U = 5 eV for the Fe-3d electrons.[16,17]
3. Results and discussion
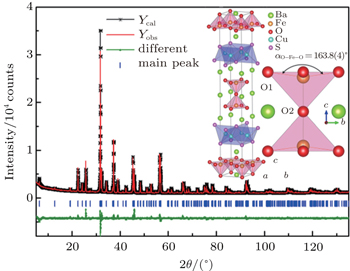
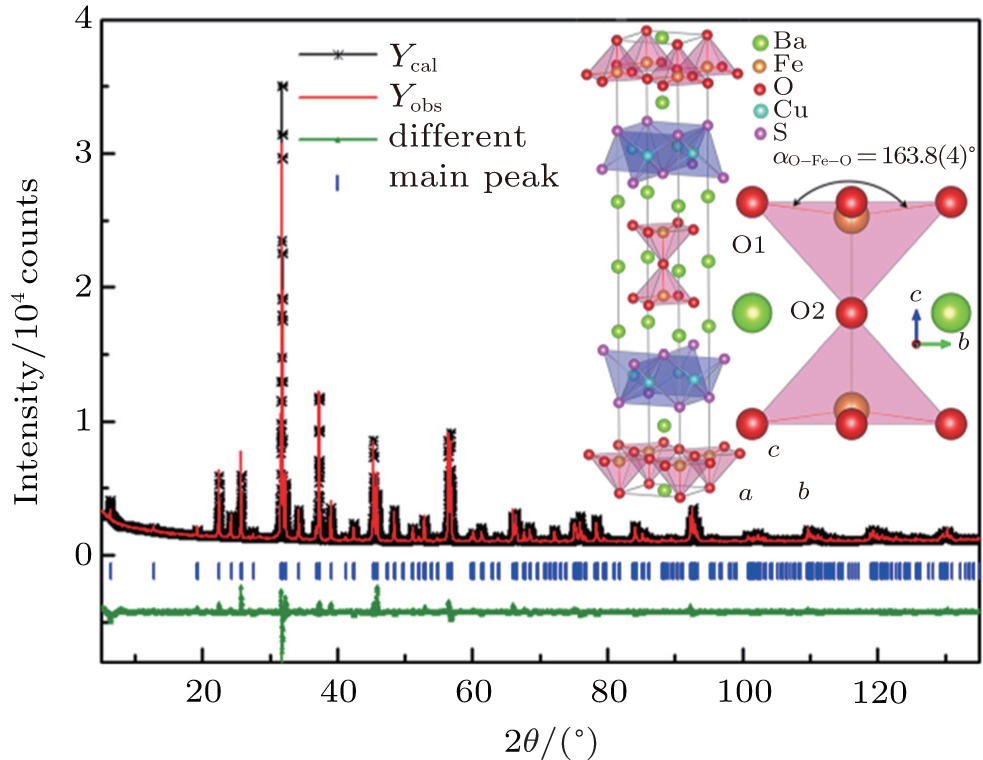
The room temperature PXRD pattern of the Ba3Fe2O5Cu2S2 sample was shown in Fig. 1. All diffraction peaks of Ba3Fe2O5Cu2S2 can be well indexed on a tetragonal cell (space group I4/mmm) with lattice parameters a = 3.9995(1) Å and c = 27.6873(3) Å. The structure of Sr3Fe2O5Cu2S2[18] was used as a starting model for the Rietveld refinement against the raw data. The agreement factors of final Rietveld refinement are Rp = 3.47%, Rwp = 5.02%, and χ2 = 3.57, indicating the correct adoption of the starting model. The refinement results are summarized in Table 1. The final crystal structure is shown in the inset of Fig. 1, which is isostructural to Sr3Fe2O5Cu2S2. The structure of Ba3Fe2O5Cu2S2 is built up by alternatively stacking [Cu2S2]2− layers and iron perovskite oxide [(FeO2)(BaO)(FeO2)]2− layers along the c axis that are separated by barium ions. It is worth noting that the Fe3+ cation is fivefold coordinated by a square-pyramidal arrangement of oxygen rather than a square-planar arrangement. For all the known layered CuCh-based compounds with perovskite oxide sheets, the cations in perovskite oxide sheets locate a square pyramidal or a square planar oxygen coordination geometry. From the bond valence arguments, we inferred that the cation locates a square pyramidal oxygen coordination geometry if its valence is +3;[3,18–21] otherwise the cation locates a square-planar oxygen coordination geometry if its valence is less than +3.[20,22–25] The relationship between cation positions and the bond valence was also found in YBa2Cu3O7−x, where the Cu2+ and Cu3+ cations preferentially occupy square pyramidal and square planar sites, respectively.[26]
Fig. 1. PXRD pattern collected from the Ba3Fe2O5Cu2S2 sample and the Rietveld refinement profiles.
Table 1.
Table 1.
Table 1.
Room-temperature crystallographic data for Ba3Fe2O5Cu2S2.
.
Ba3Fe2O5Cu2S2
Crystal system
Tetragonal
Space group
I4/mmm (No. 139)
a/Å
3.9995(1)
c/Å
27.6873(3)
V/Å3
442.884(8)
Atomic positions
x
y
z
Uiso/100 Å2
Ba1(2b)
1/2
1/2
0
0.33(8)
Ba2(4e)
1/2
1/2
0.14326(5)
0.91(6)
Fe(4e)
0
0
0.07020(16)
1.55(11)
O1(8g)
1/2
0
0.0805(3)
0.887(3)
O2(2a)
0
0
0
0.633(5)
Cu(4d)
1/2
0
1/4
0.830(5)
S(4e)
0
0
0.19982(19)
0.89(20)
Coordination
αO1−Fe−O1/(°)
163.8(4)
aS−Cu−S/(°)
110.419
Agreement factors
Rp = 0.0347
Rwp = 0.0502
Table 1.
Room-temperature crystallographic data for Ba3Fe2O5Cu2S2.
.
Figure 2(a) shows the temperature-dependent susceptibility χ(T) of Ba3Fe2O5Cu2S2 measured under a magnetic field strength of 2000 Oe (1 Oe = 79.5775 A·m−1). The χ(T) curve in high-T range of 146 K–290 K can be fitted well by the Curie–Weiss law χ = χ0 + c/(T − θ), where C and θ are the Curie constant and the Weiss temperature, respectively. The fitted parameters χ0, C, and θ are 0.0125 emu·Oe−1·mol−1, 9.18419 emu·K·Oe−1·mol−1 and –504 K, respectively. The effective magnetic moment μeff is determined to be 6.05(1) μB, which is in agreement with the value of 5.92 μB expected for the high spin d5 configuration of Fe3+.[27] The negative value of θ indicates that the dominant magnetic interactions in the compound are antiferromagnetic (AFM). The sharp peak at 121 K indicates an AFM transition. Also, the dependence of isotemperature magnetization on magnetic field H shows that magnetizationM is nearly proportional to H even down to 10 K, with minor hysteresis possibly due to trace impurities that undetectable in PXRD. The Curie–Weiss-like upturn at low temperatures is presumably due to the paramagnetic impurities as in Ba2CoO2Ag2Se2.[28]
Fig. 2. (a) Zero field cooling (ZFC) and field cooling (FC) χ(T) data taken in an applied field of 2 kOe. The fit to the Curie–Weiss law is shown as a green line. Inset: the M–H curves at 10 K. (b) Temperature dependence of specific heat Cp(T) of Ba3Fe2O5Cu2S2 from 10 K to 300 K.
The heat capacity versus T at constant pressure Cp at H = 0 was plotted in Fig. 2(b). There is no clear anomaly at AFM transition temperature TN of 121 K, despite the relatively sharp magnetic ordering transition observed by magnetization. Analyses of Cp(T) curves commonly yield magnetic entropies above TN smaller than expected for the paramagnetic state, mostly due to short range magnetic correlations at T > TN and the difficulty of separating the phonic contrubution from the spin-wave specific heats at T < TN. This phenomenon was also observed in LaCrSb3[29] and Sr2Mn3As2O2.[30] As shown in the inset of Fig. 2(b), Cp(T)/T versus T2 shows a perfect linear characteristic at low temperature (10 K∼15 K). It is fitted by the expression Cp/T = γ + βT2, where γ is the Sommerfeld coefficient. The fitted parameters β and γ are 0.0024 J·mol−1·K−4 and 0.1205 J·mol−1·K−2, respectively. According to the formula ΘD = (12π4NR/β)1/3, the Debye temperature ΘD is estimated to be about 113 K.
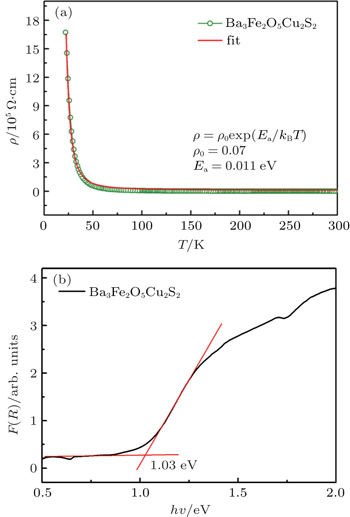
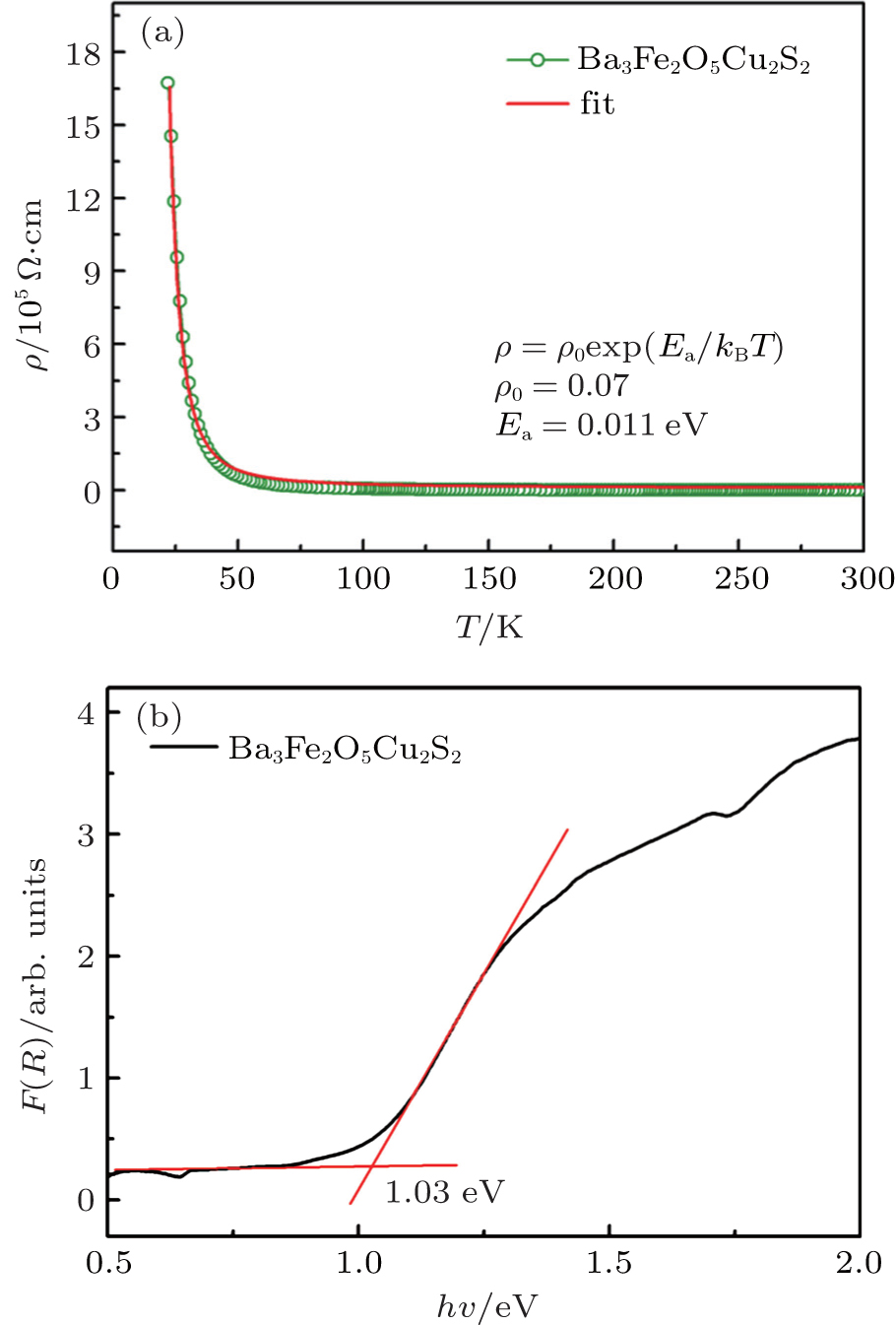
Figure 3(a) gives the variation of resistivity with temperature for Ba3Fe2O5Cu2S2. It exhibits a semiconducting behavior from 55 K to 300 K. The ρ(T) obeys the thermally activated behavior ρ = ρ0 exp (Ea/kBT), where Ea is the activation energy. The obtained Ea is 0.011 eV, which is much smaller than that of Ca2FeO3CuS (0.19 eV).[19] For a magnetic semiconductor, electrical conduction results mainly from electron-hopping between adjacent spin sites. Thus, the longer Fe–Fe and Fe–O distances in Ba3Fe2O5Cu2S2 result in a weaker overlapping integral between the electron-wave-function at the adjacent Fe3+ sites for hopping conduction, which explains the small electrical conductivity observed than that of Ca2FeO3CuS.
Fig. 3. (a) Temperature dependence of the resistivity ρ(T) of the Ba3Fe2O5Cu2S2 with H = 0. The red line is the fitting results of ρ(T) using thermal activation model. (b) UV-vis-NIR diffuse reflectance spectrum of Ba3Fe2O5Cu2S2.
The UV-vis-NIR diffuse reflectance spectra of Ba3Fe2O5Cu2S2 were displayed in Fig. 3(b) In the F(R) versus hν plots, the absorption edge can be deduced via the straightforward extrapolation method. The optical band gap is estimated to be about 1.03 eV, which is consistent with the gray-black color of the material. The optical band gap of Ba3Fe2O5Cu2S2 is much larger than its activation energy value of 0.011 eV deduced from resistivity. The disparity between the two energy values is understandable since the activation energy Ea extracted from the resistivity of the polycrystalline samples may be resulted from shallow impurity levels located below (above) conduction (valence) band.
The electrical properties and bandgaps of typical compounds with and without magnetic atoms were summarized and compared in Table 2, where the majority of reported Cu-based oxychalcogenides are semiconductors. A few compounds present metallic behavior because less than two electrons are transported to [Cu2Ch2]2− layer so that their valance bands are not fully filled.[32] It is found that the bandgaps of the titled compound and the Sr2MO2Cu2Se2 (M = Co, Mn) are much less than the typical band-gaps 2 eV–3 eV of other reported oxychalcogenide semiconductors that without 3d electrons.[1–3,9,10,33] Thus, it is speculated that the magnetic atoms in the compounds have a significant effect on the electronic structure.
Table 2.
Table 2.
Table 2.
Several typical CuCh-based compounds and their electrical properties.
Several typical CuCh-based compounds and their electrical properties.
.
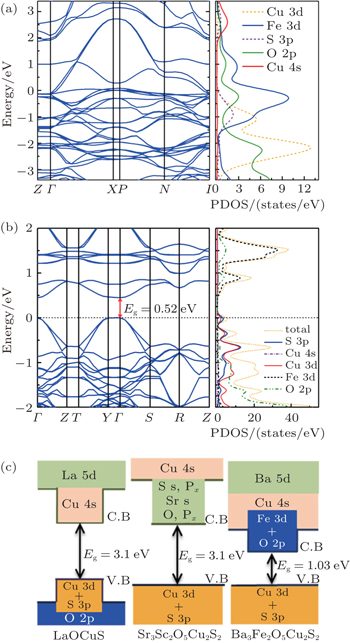
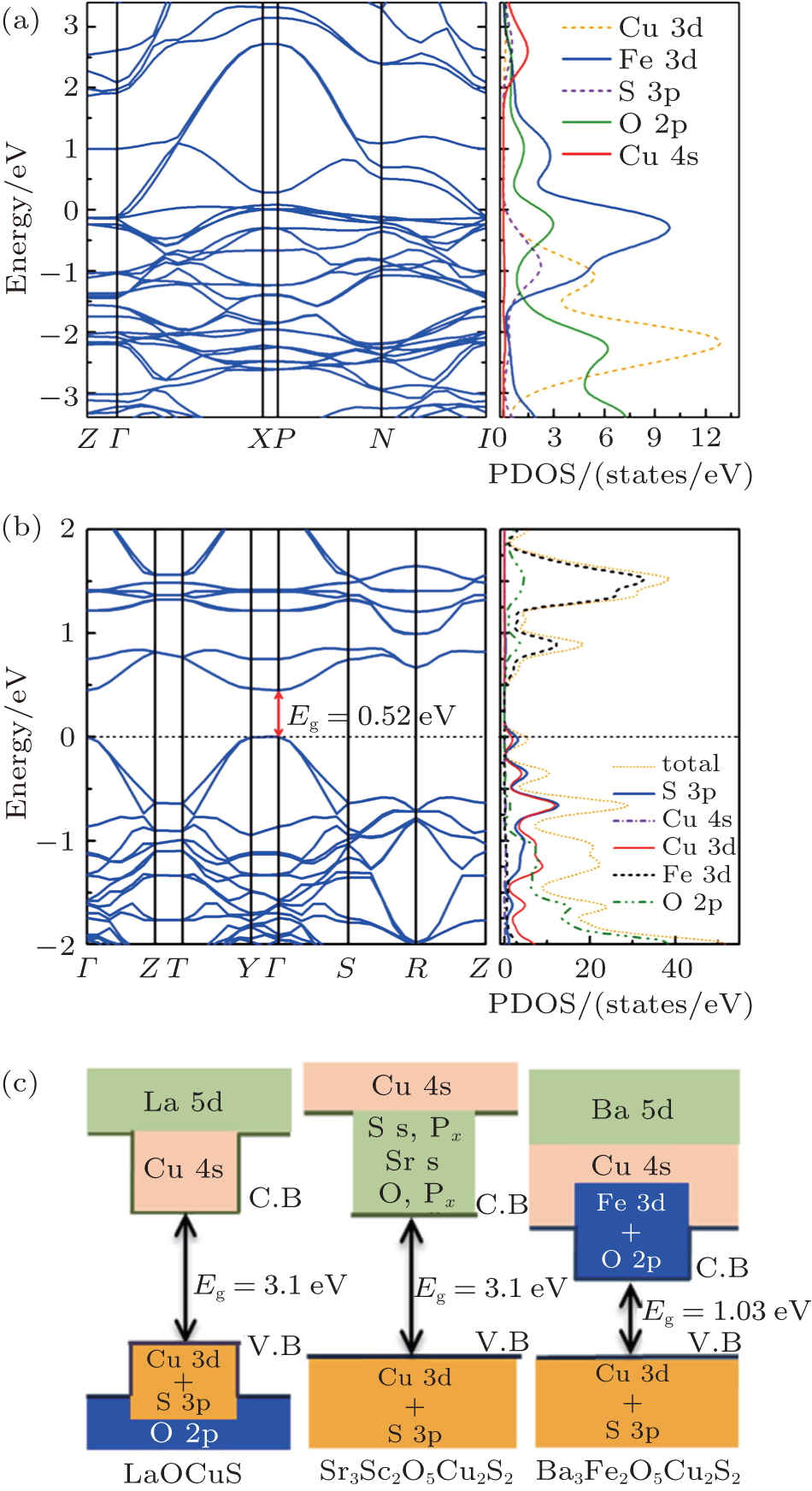
Electronic structures of the titled compound were calculated based on the first principles calculations considering the two cases of the nonmagnetic structure and the spin-polarized structure. A nonmagnetic calculation was first performed where the spins of the magnetic atoms are not considered and the calculated band structure along the high-symmetry k lines was showed in Fig. 4(a). The bands located from 0 eV to 2 eV basically come from the hybridization of O 2p and Fe 3d states The bands located from –2 eV to 0 eV are contributed by Cu 3d and S 3p states besides the O 2p and Fe 3d states, while the Cu 4s bands distribute above 2 eV. The obtained band structure for Ba3Fe2O5Cu2S2 exhibits a metal feature, which is inconsistent with the previous result of resistivity measurements.
Fig. 4. Band structure (left) and total/atom resolved partial density of states (PDOS) (right) near the Fermi energy for (a) non-magnetic Ba3Fe2O5Cu2S2 and (b) antiferromagnetic Ba3Fe2O5Cu2S2 with U = 5 eV. (c) The schematic diagrams of band structure for LaOCuS, Sr3Sc2O5Cu2S2 and antiferromagnetic Ba3Fe2O5Cu2S2, respectively.
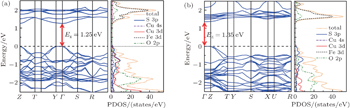
Then, a spin-polarized calculation was performed by using LDA + U method with U = 5 eV. A G-type AFM structure was adopted for Ba3Fe2O5Cu2S2 as reported in Sr3Fe2O5Cu2S2.[34] As shown in Fig. 4(b), both the minimal-energy state in the conduction bands and the maximal-energy state in the valence bands are located at the Γ point (k = 0), indicating that Ba3Fe2O5Cu2S2 is a direct band-gap semiconductor with a bandgap of 0.52 eV. The underestimation relative to the experimentally measured optical band gap of 1.03 eV is on account of the well-known problem that LDA + U calculations always give underestimated band gaps.[35] The spin-polarized atom-resolved density of states are shown in the right panel of Fig. 4(b). The valence band maximum, which is mostly contributed by the Cu 3d states and S 3p states, is similar to quaternary oxychalcogenides[1,10,33] and multilayered mixed-metal oxychalcogenides,[2,3,9] while the conduction band minimum is different even between the isostructural compounds. As schematically diagrammed in Fig. 4(c), the CBM of Ba3Fe2O5Cu2S2 is dominated by the Fe 3d and O 2p states, while the CBM of the isostructural compound Sr3Sc2O5Cu2S2 is dominated by sulfur s and pz states, Sr s states and O px states,[36] and the CBM of LaOCuS is occupied by Cu 4s orbitals.[37] As shown in Fig. 5, the CBM of Sr3Fe2O5Cu2S2 and Ca2FeO3CuS compounds are also comprised of the Fe 3d/O 2p states through the first principles calculations. They are direct band-gap semiconductors with a bandgap of 1.25 eV and 1.35 eV, respectively. The CBM that are comprised of the Fe 3d/O 2p states lie at much lower energy compared with nonmagnetism compound Sr3Sc2O5Cu2S2, which result in a bandgap narrowing. The band gaps can be divided into two regimes in terms of gap values: the layered CuCh-based compounds without magnetic cations fall in Regime I (band gap range from 2 eV to 3 eV); the layered CuCh-based compounds with magnetic cations fall in Regime II (band gap range from 0 eV to ∼1 eV). Therefore, the magnetism in perovskite oxide sheets predominantly affects the conduction band minimum and results in a much narrower band gap.
Fig. 5. Band structure (left) and total/atom resolved partial density of states (PDOS) (right) near the Fermi energy for antiferromagnetic Sr3Fe2O5Cu2S2 (a) and Ca2FeO3CuS (b) with U = 5 eV.
4. Conclusion
In summary, a new layered oxychalcogenides compound Ba3Fe2O5Cu2S2 was synthesized and the underlying bandgap narrowing mechanism is revealed by experimental and DFT calculations. Refinement of powder x-ray diffraction data shows that its structure is built up by alternatively stacking [Cu2S2]2 layers and iron perovskite oxide [(FeO2)(BaO)(FeO2)]2− layers along the c axis that are separated by barium ions, with Fe3+ cation locates in a square-pyramidal oxygen coordination geometry. From the bond valence arguments we inferred that in layered CuCh-based oxychalcogenides with perovskite oxide sheets the +3 cationsoccupy square pyramidal sites while the cations with less than +3 locate square planar sites. The optical bandgap of Ba3Fe2O5Cu2S2 is about 1.03 eV, much smaller than the reported nonmagnetic oxychalcogenides. The magnetic susceptibility and transport measurements indicate that Ba3Fe2O5Cu2S2 is an AFM ordering semiconductor with a Néel temperature of 121 K. Electronic structures of a nonmagnetic calculations present metallic behavior, while for a spin-polarized calculation on AFM magnetic structure the results show that Ba3Fe2O5Cu2S2 has a considerable bandgap opened and CBM that are comprised of the Fe 3d/O 2p states lie at much lower energy compared with isostructural Sr3Sc2O5Cu2S2, which result in a bandgap narrowing.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 , Guo Liwei1, Shen Shijie1, Lin Zhiping1, Chen Xiaolong1, 2]
, Guo Liwei1, Shen Shijie1, Lin Zhiping1, Chen Xiaolong1, 2]