Zhao Qing, Yin Yun-Yu, Dai Jian-Hong, Shen Xi, Hu Zhi-Wei, Yang Jun-Ye, Wang Qing-Tao, Yu Ri-Cheng, Li Xiao-Dong, Long You-Wen. A-site ordered perovskite CaCu3Cu2Ir2O12−δ with square-planar and octahedral coordinated Cu ions. Chinese Physics B, 2016, 25(2): 020701
Permissions
A-site ordered perovskite CaCu3Cu2Ir2O12−δ with square-planar and octahedral coordinated Cu ions
Zhao Qing1, 2, Yin Yun-Yu2, Dai Jian-Hong2, Shen Xi2, Hu Zhi-Wei3, Yang Jun-Ye2, Wang Qing-Tao1, 4, †, , Yu Ri-Cheng2, Li Xiao-Dong5, Long You-Wen2, 6, ‡,
College of Physics, Qingdao University, Qingdao 266071, China
Beijing National Laboratory for Condensed Matter Physics, Institute of Physics, Chinese Academy of Sciences, Beijing 100190, China
Max-Planck Institute for Chemical Physics of Solids, Nöthnitzer Straße 40, 01187 Dresden, Germany
Key Laboratory of Photonics Materials and Technology in Universities of Shandong (Qingdao University), Qingdao 266071, China
Beijing Synchrotron Radiation Facility, Institute of High Energy Physics, Chinese Academy of Sciences, Beijing 100049, China
Collaborative Innovation Center of Quantum Matter, Beijing 100190, China
† Corresponding author. E-mail: wangqt@qdu.edu.cn
‡ Corresponding author. E-mail: ywlong@iphy.ac.cn
Project supported by the National Basic Research Program of China (Grant No. 2014CB921500), the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB07030300), and the National Natural Science Foundation of China (Grant No. 11574378).
Abstract
Abstract
A novel CaCu3Cu2Ir2O12−δ polycrystalline sample was synthesized at 8 GPa and 1373 K. Rietveld structural analysis shows that this compound crystallizes in an -type A-site ordered perovskite structure with space group Im-3. X-ray absorption spectra reveal a +2-charge state for both the square-planar and octahedral coordinated Cu ions, and the valence state of Ir is found to be about +5. Although the A-site Ca and the A′-site Cu2+ are 1:3 ordered at fixed atomic positions, the distribution of B-site Cu2+ and Ir5+ is disorderly. As a result, no long-range magnetic ordering is observed at temperatures down to 2 K. Electrical transport and heat capacity measurements demonstrate itinerant electronic behavior. The crystal structure is stable with pressure up to 35.7 GPa at room temperature.
A-site ordered quadruple perovskites with a chemical formula of have attracted much attention. In this ordered system, both the A′ and B sites can accommodate transition-metal ions, giving rise to multiple magnetic and electrical interactions such as A′–A′, B–B, and A′–B interactions. As a consequence, a series of intriguing physical properties have been observed in A-site ordered perovskite oxides. For example, ferromagnetic phase transitions caused by the A′-site Cu2+ ions were found in CaCu3Ge/Sn4O12,[1,2] whereas in CaCu3Ti4O12 the spin ordering of Cu2+ leads to an antiferromagnetic transformation.[3,4] More interesting, temperature/pressure-induced intermetallic charge transfer occurs between the A′-site Cu and the B-site Fe in La/BiCu3Fe4O12, resulting in sharp variations in lattice, charge and spin degrees of freedom.[5–7] When the A-site La/Bi was replaced by Ca, charge disproportionation instead of charge transfer took place in CaCu3Fe4O12.[8] Unlike La/Bi/CaCu3Fe4O12, colossal negative thermal expansion was shown in SrCu3Fe4O12 in a wide temperature window.[9]
The A-site ordered perovskite usually crystallizes to a cubic crystal structure with space group Im-3 as shown in Fig. 1. In this symmetry, the A and A′ sites are 1:3 ordered to occupy different atomic positions 2a (0, 0, 0) and 6b (0, 0.5, 0.5), respectively. The B site is fixed at 8c (0.25, 0.25, 0.25), and the O is located at 24g (0, y, z). Compared to the simple ABO3 perovskite where the 12-fold coordinated A site is often occupied by alkali metal, alkaline earth, and/or lanthanide cations,[10–12] in the present ordered case, three quarters of the A-sites are substituted by a transition metal A′ with smaller ionic radius. Therefore, the BO6 octahedra are heavily tilted (typically, ∠B–O–B ≈ 140°) and the A′-site transition metal forms a square-planar A′O4 unit.[13] Since the coordination environment can play an important role for the electronic configuration of transition metals, the A-site ordered perovskite provides a unique material system in which to study the effects of coordination on material physical properties. In this paper, we report preparing for the first time the A-site ordered perovskite CaCu3Cu2Ir2O12−δ (CCCIO) with Cu ions occupying the square-planar A′O4 units as well as the octahedral BO6 polyhedra. Structural and x-ray absorption analyses show that the charge combination is with δ ≈ 1, and the B-site Cu2+ and Ir5+ ions are distributed in a disordered manner. No long-range magnetic ordering is observed at temperatures down to 2 K.
Fig. 1. Crystal structure of A-site ordered perovskite with space group Im-3. The A′-site transition metal forms a square-planar A′O4 unit, whereas the B-site transition metal forms an octahedral coordinated BO6 unit.
2. Experiment
Polycrystalline CCCIO was prepared under high-pressure, high-temperature conditions by using a cubic-anvil-type high-pressure apparatus. Stoichiometric amounts of high-purity (> 99.9%) CaO, CuO, IrO2 powders were used as starting materials, and excess KClO4 was used as oxygen source. These reactants were mixed thoroughly in an agate mortar. The mixed powders were sealed into a gold capsule with 2.8 mm in diameter and 4.0 mm in length and then treated at 8 GPa and 1373 K for half an hour. When the heating time was finished, the sample was quenched to room temperature, and the pressure was released slowly. The residual KCl in the final product was washed out by deionized water.
The sample purity and crystal structure were examined by powder x-ray diffraction (XRD) using a Rigaku x-ray diffractometer with Cu–Kα radiation (40 kV, 300 mA). The XRD data were collected in the angle range from 10° to 130° with steps of 0.02° and analyzed by the Rietveld refinement using the GSAS program.[14] A Philips CM200 transmission electron microscope (TEM) with a field emission gun operated at 200 keV was used for the select-area electron diffraction (SAED) investigation. The valence states of Cu and Ir were identified by x-ray absorption spectra (XAS) performed at the National Synchrotron Radiation Research Center in Taiwan. The soft XAS at the Cu–L3 edges were measured with total electron yield at the beamline of BL08B. The hard XAS at the Ir–L3 edges were measured in the transmission geometry at the BL07A beamline.
Magnetic susceptibility and magnetization were measured using a Quantum Design superconducting quantum interference device magnetometer. The temperature dependence of the magnetic susceptibility was measured at 0.1 T over a range from 2 K to 300 K. The field dependence of the magnetization was measured at several temperatures and under fields from −7 T to 7 T. Temperature dependence of resistivity and specific heat were measured by using a Quantum Design physical property measurement system.
High-pressure synchrotron x-ray diffraction (SXRD) at room temperature was performed at Beijing Synchrotron Radiation Facility. The wavelength used for this experiment was λ = 0.6199 Å. The powder sample was pressurized using a diamond anvil cell (DAC) with a pair of 300-μm culet. Silicone oil was used as pressure transmitting medium and the pressures were calibrated on the basis of the ruby fluorescence method.[15] The SXRD data were analyzed using the Rietveld refinement program GSAS.
3. Results and discussion
Figure 2 shows the powder XRD pattern measured at room temperature as well as the Rietveld refinement results of CCCIO. The XRD pattern can be well fitted based on the A-site ordered perovskite structure model with space group Im-3. This means that the B-site Cu and Ir ions are arranged randomly over the available octahedral sites. Furthermore, when the SAED is performed, one can find no evidence of B-site ordering. As an example, the inset of Fig. 2 shows the SAED pattern obtained along the zone axis. The absence of diffraction spots with h + k + l = odd confirms the B-site disorder.[16,17] Therefore, the present CCCIO crystallizes in the same crystal structure as that of CaCu3Cr2Ru2O12, wherein the B-site Cr3+ and Ru5+ are also disordered.[18,19] The refined structural parameters of CCCIO are listed in Table 1. The lattice parameter of CCCIO (7.4481 Å) is slightly less than that of CaCu3Ir4O12 (7.4738 Å) due to partial substitution of Ir by Cu ions at the B site.[20] According to the A′-site Cu–O bond lengths, the calculated bond valence sum (BVS) of Cu is 1.92, indicating a Cu2+ state in this square-planar site.
Fig. 2. XRD pattern and the Rietveld refinement profile of CaCu3Cu2Ir2O12−δ. The observed (circles), calculated (red line), and difference (blue line) patterns are shown. The ticks show the positions of the Bragg reflections with space group Im-3. The inset shows the SAED image along the zone axis.
Table 1.
Table 1.
Table 1.
Refined structure parameters of CaCu3Cu2Ir2O12−δ at room temperature. Space group: Im-3; Atomic sites: Ca 2a (0, 0, 0), Cu 6b (0, 0.5, 0.5), Cu/Ir 8c (0.25, 0.25, 0.25), O 24g (0, y, z).
.
Parameter
CCCIO
a/Å
7.44817(4)
Oy
0.181
Oz
0.307
Uiso (Ca)/(100 × Å2)
0.04(6)
Uiso (Cu)/(100 × Å2)
2.14(5)
Uiso (Cu/Ir)/(100 × Å2)
0.5(2)
Uiso (O)/(100 × Å2)
0.04(1)
Ca–O/(×12 Å)
2.654(4)
Cu–O/(×4 Å)
1.970(7)
(×4) (Å)
2.777(1)
(×4) (Å)
3.297(5)
Cu/Ir–O/(×6 Å)
1.977(7)
∠O–Cu–O/(°)
93.6(7)
∠O–Cu/Ir–O/(°)
90.7(5)
∠Cu/Ir–O–Cu/Ir/(°)
140.6(1)
∠Cu–O–Cu/Ir/(°)
109.5(3)
Rwp/%
7.15
Rp/%
5.22
Table 1.
Refined structure parameters of CaCu3Cu2Ir2O12−δ at room temperature. Space group: Im-3; Atomic sites: Ca 2a (0, 0, 0), Cu 6b (0, 0.5, 0.5), Cu/Ir 8c (0.25, 0.25, 0.25), O 24g (0, y, z).
.
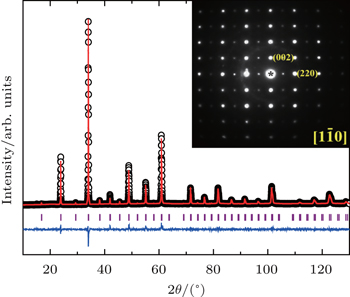
X-ray absorption spectrum at the transition-metal L3 edge is element-specific and sensitive to the valence state as well as the local environment of the transition metal. The valence states of Cu and Ir in our CCCIO are therefore determined by XAS method. Figure 3(a) shows the Cu–L3 XAS of CCCIO together with Ca0.83Cu2.34+O2 and . The CaCu3Ti4O12 is used as a pure divalent reference for the A′-site Cu ion with a CuO4 square-planar coordination, which has the same local environment as CCCIO for Cu ions at the A′ site. One can see a sharp peak at the Cu–L3 edge in CaCu3Ti4O12 and CCCIO owing to the dipole transition 2p63d9 → 2p53d10,[21] reflecting the same Cu2+ valence state. However, there is an energy shift about 0.29-eV higher from CaCu3Ti4O12 (931.37 eV) to CCCIO (931.66 eV). That can be understood by the different local environment of Cu. The Cu2+ ions in CCCIO have square-planar coordination and octahedral coordination at the same time. The Ca0.83CuO2 is adopted as another reference with mixed Cu2+ and Cu3+ states.[22] The energy position of the Cu2+ related peak in the mixed-valence Ca0.83CuO2 shifts by about 0.25 eV toward the higher energy side relative to CaCu3Ti4O12. Because the Cu ions in Ca0.83CuO2 and CaCu3Ti4O12 have a similar local square-planar coordination, but the Cu–O network in the former is edge-shared, whereas the CuO4 units in CaCu3Ti4O12 are separated from each other. For Ca0.83CuO2, the sharp Cu3+-related peak at 933.57 eV in Fig. 3(a) is assigned to 3d9L, where L stands for hole at O-2p states. It is only observed for an edge-shared or an isolated Cu–O network,[23,24] while the Cu3+ion-related spectral feature with a corner-shared Cu–O network shows only a broad shoulder above the Cu2+-related peak.[25,26] Thus, we can conclude that our sample is free from Cu3+ ions: all the Cu ions in CCCIO are Cu2+ states.
Fig. 3. XAS of (a) Cu–L3 edge, (b) Ir–L3 edge of CaCu3Cu2Ir2O12−δ. The XAS spectra of some related references are also shown for comparison.
The XAS of CCCIO along with Sr3ZnIr4+O6 and Sr3NaIr5+O6 are shown in Fig. 3(b). These two references with different Ir valence states have the same local environment (IrO6 octahedra).[27] By comparison, the white line in the Ir–L3 edge of CCCIO lies at the same energy position as that of Sr3NaIrO6 with Ir5+, and deviates from Sr3ZnIr4+O6. As a result, XAS measurements prove the presence of Ir5+ in CCCIO. Having determined the A′-site Cu2+, the B-site Cu2+ valence states and the B-site Ir5+ valence states, we conclude that CCCIO is nonstoichiometric, in connection with the slight oxygen deficiency.
The magnetic susceptibility of CCCIO as a function of temperature is shown in Fig. 4(a). With temperature decreasing down to 2 K, no long-range magnetic phase transition is observed. Above 100 K, the susceptibility is nearly temperature-independent, probably suggesting metallic Pauli paramagnetic behavior. The Curie-like upturns observed at low temperatures may originate from a small amount of impurity phases, which are undetectable by XRD.[28] Figure 4(b) shows the field dependence of isothermal magnetization measured at different temperatures. The linear magnetization behaviors are consistent with the paramagnetic feature as determined from the magnetic susceptibility measurements.
Fig. 4. (a) Temperature dependence of magnetic susceptibility of CaCu3Cu2Ir2O12−δ measured at zero-field cooling (ZFC) and field cooling (FC) modes with applied field 0.1 T. (b) Field dependence of the magnetization at different temperatures. The unit 1 Oe = 79.5775 A·m−1.
As is well known, the Ir5+ ions (5d4) has a Jeff = 0 singlet ground state in the strong spin–orbit coupling limit.[29] This means that the B-site Ir5+ in the present CCCIO is nonmagnetic in nature. Although it is possible for the B-site Cu2+ (3d9) to take part in spin ordering as observed in Sr2CuIrO6,[30] the random distribution between Cu2+ ions and the nonmagnetic Ir5+ is unfavorable for the formation of long-range magnetic ordering at the B site in CCCIO. In addition, in CaCu3Ge4O12 and CaCu3Sn4O12, the A′-site Cu2+ ions can induce a ferromagnetic phase transition via the Cu–Cu direct interactions. On the other hand, in CaCu3Ti4O12, the d0-orbits of Ti4+ ions are found to contribute to the antiferromagnetic ordering through the Cu2+–O–Ti4+–O–Cu2+ super-superexchange pathway.[31–34] In our CCCIO, since the d-orbits of the B-site transition metals are disordered, no long-range spin ordering can be formed in the whole temperature range we measured (2 K–300 K).
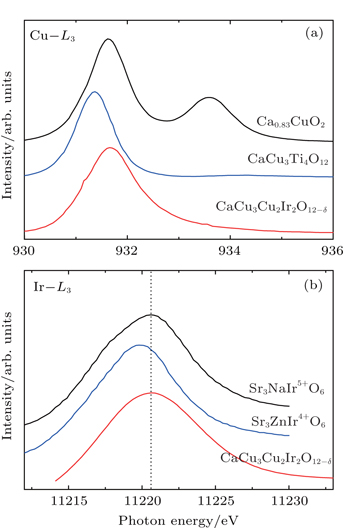
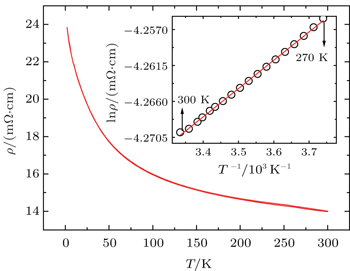
Figure 5 shows the temperature dependence of resistivity for CCCIO. With decreasing temperature, the resistivity increases slightly from 14 mΩ·cm at 300 K to 24 mΩ· cm at 2 K. As shown in Fig. 1, the A′O4 units in the A-site ordered perovskite are separated from each other in space, the electrical transport therefore should be dominated by the corner-sharing BO6 octahedra. Considering that the resistivity of CCCIO is measured on a polycrystalline pellet, where the grain-boundary scattering plays an important role,[35] the observed small, and only slightly temperature-dependent, resistivity values may imply the delocalized electronic feature for the B-site disordered Ir5+ and Cu2+ ions. Actually, in Sr2Fe1−xCuxMoO6 with Cu2+ ions, and in Ba0.5IrO3 with Ir5+ ions, metallic electrical conductivities were observed.[36,37] The resistivity data of CCCIO in 270 K–300 K can be well fitted by using the thermal activation model with the formula ρ = ρ0eEA/KT as shown in the inset of Fig. 5. Here EA and K stand for thermal activation energy and the Boltzmann constant, respectively. The fitting gives EA= 3.05(2) meV. This very small and probably physically meaningless activation energy may also reflect the delocalized behavior of the polycrystalline CCCIO.
Fig. 5. Temperature dependence of resistivity of CaCu3Cu2Ir2O12−δ. The inset shows the fitting result using the thermal activation model. The open circles represent experimental data, and the red line is the fitting result.
To further understand the transport properties of CCCIO, heat capacity (CP) was measured as shown in Fig. 6. In the temperature region we measured (2 K–200 K), we do not find any anomaly in CP, in agreement with the absence of any long-range spin ordering. The inset of Fig. 6 presents the low-temperature fitting result using the formula CP/T = γ + βT2, where the γT and βT3 terms describe the electron and lattice contributions to the total heat capacity, respectively. The obtained coefficient γ is equal to 0.211(8) J·mol−1·K−2, which is larger than that of β (0.0039(3) J·mol−1·K−4) by two orders of magnitude. Therefore, the electron contribution dominates the low-temperature heat capacity, confirming the intrinsic itinerant electronic behavior of CCCIO, related to the B-site-disordered Cu2+ and Ir5+ ions.
Fig. 6. Heat capacity as a function of temperature for CaCu3Cu2Ir2O12−δ. The inset shows the fitting result described in the text. The open circles represent experimental data, and the red curve is the fitting result.
In order to investigate structural stability of CCCIO under high pressure, the SXRD was carried out at room temperature. Figure 7(a) shows some representative SXRD patterns collected at different pressures. As the pressure increases, one can find that all the diffraction peaks systematically shift towards higher 2θ values as expected from the unit cell volume contraction. No visible structural phase transition is observed in the pressure region we applied, suggesting that the crystal structure of CCCIO is stable up to 35.7 GPa. Figure 7(b) shows the related pressure dependence of lattice constant. It monotonously decreases with increasing pressure without any profound change.
Fig. 7. (a) SXRD patterns of CaCu3Cu2Ir2O12−δ at selected pressures and room temperature. The asterisks stand for the diffraction peak originating from KCl impurity phase. (b) Pressure dependence of lattice constant of CaCu3Cu2Ir2O12−δ. The error bars are located within the data symbols.
4. Summary
In summary, polycrystalline CaCu3Cu2Ir2O12−δ has been prepared for the first time, by a high-pressure, high-temperature method. The Rietveld refinement reveals that this compound crystallizes in an A-site ordered but B-site disordered perovskite structure with space group Im-3. Both BVS calculation and XAS confirm the charge states to be Cu2+ and Ir5+. Because of the disordered distribution of the B-site Ir5+ and Cu2+, no long-range magnetic phase transition is observed in the whole temperature region we measured (2 K–300 K). The susceptibility approximately shows a temperature-independent Pauli paramagnetic behavior. Based on the analysis of resistivity data, we infer delocalized electrical behavior for the present CCCIO. Furthermore, the itinerant electronic feature is confirmed by heat capacity analysis, with the electron contribution dominating the low-temperature heat capacity. High-pressure SXRD measurement does not show any evidence for possible structural phase transition, revealing the structural stability of CCCIO with pressure up to 35.7 GPa at room temperature.
A-site ordered perovskite CaCu3Cu2Ir2O12−δ with square-planar and octahedral coordinated Cu ions
[Zhao Qing1, 2, Yin Yun-Yu2, Dai Jian-Hong2, Shen Xi2, Hu Zhi-Wei3, Yang Jun-Ye2, Wang Qing-Tao1, 4, †, , Yu Ri-Cheng2, Li Xiao-Dong5, Long You-Wen2, 6, ‡, ]


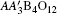




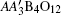



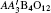









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 , Yu Ri-Cheng2, Li Xiao-Dong5, Long You-Wen2, 6, ‡,
, Yu Ri-Cheng2, Li Xiao-Dong5, Long You-Wen2, 6, ‡,