

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
High-throughput theoretical design of lithium battery materials
[Ling Shi-Gang , Gao Jian , Xiao Rui-Juan †,  , Chen Li-Quan ]
, Chen Li-Quan ]
, Chen Li-Quan ]
|
|


† Corresponding author. E-mail:
Project supported by the National Natural Science Foundation of China (Grant Nos. 11234013 and 51172274) and the National High Technology Research and Development Program of China (Grant No. 2015AA034201).
The rapid evolution of high-throughput theoretical design schemes to discover new lithium battery materials is reviewed, including high-capacity cathodes, low-strain cathodes, anodes, solid state electrolytes, and electrolyte additives. With the development of efficient theoretical methods and inexpensive computers, high-throughput theoretical calculations have played an increasingly important role in the discovery of new materials. With the help of automatic simulation flow, many types of materials can be screened, optimized and designed from a structural database according to specific search criteria. In advanced cell technology, new materials for next generation lithium batteries are of great significance to achieve performance, and some representative criteria are: higher energy density, better safety, and faster charge/discharge speed.
The development of new materials is of major technological importance for almost any application. The traditional way to discover a new material is to produce and test samples one by one based on empirical information about crystal structures and other material properties. [ 1 ] The numerous combinations of elements, compositions and crystal structures make the search for needed materials an arduous task, and the time from discovery of a new material to its use in commercial products can be 20 years or more. [ 2 ] To accelerate the pace of materials discovery, a strategy originally developed in pharmacy labs, high-throughput combinatorial screening, [ 3 – 5 ] has been more widely adopted to search for new materials or improve materials already in use. Rather than create and test materials one by one, the high-throughput methods synthesize and characterize new materials through thousands of samples in parallel, greatly speeding up the identification of valuable materials. [ 6 ] Starting from the significant work by Xiang et al. for discovering superconductors through combinatorics in 1995, [ 7 ] the high-throughput method has been recognized as effective and has been applied in biological activities, [ 11 ] and in hunting for catalysts, [ 8 – 10 ] coatings, [ 12 , 13 ] polymers, [ 14 ] fluorescence materials, [ 15 ] etc.
Over the last half century, a rapid increase of computational power and the development of theoretical methods has made quick and accurate simulations of material properties possible. As a result, high-throughput theoretical screening, analogous to high-throughput combinatorial experiments, has become a promising way to discover new materials. [ 16 ] In recent years, researchers have started to apply the highthroughput theoretical screening method to material catalogs across lots of fields. Strasser et al. performed high-throughput density functional theory (DFT) calculations on a series of ternary PtRu M ( M = transition metal) alloys to screen new anode fuel cell alloy catalysts with improved CO tolerance. [ 17 ] To search for electrocatalytic materials for the hydrogen evolution reaction (HER), Greeley et al. carried out a DFTbased high-throughput screening scheme to evaluate the activity of over 700 binary surface alloys, and BiPt was identified as an HER catalyst with better performance than Pt. [ 18 ] Andersson et al. studied the ability to combine with oxygen in the surface of 60 alloys by using density functional theory (DFT), and evaluated the materials’ catalytic activity in the conversion reaction of CO + 3H 2 →CH 4 + H 2 O, finally designing a catalyst for nickel–iron alloys. [ 19 ] By modeling the reaction of water with metal organic frameworks, Low et al. screened the alternatives for hydration of porous coordination polymers according to hydrothermal stability, based on quantum mechanical cluster calculations. [ 20 ] Wang et al. carried out high-throughput ab initio calculations of band structures for compounds from the Inorganic Crystal Structure Database (ICSD), and by evaluating the power factor of each structure, the thermoelectric properties were assessed. [ 21 ] Zhang et al. evaluated the half-Heusler compounds and pseudocubic structures as thermoelectric materials by calculating the electrical transport properties. [ 22 , 23 ] High-performance piezoelectrics were screened within the ABO3 chemical space in the perovskite structure by Armiento et al. using high-throughput DFT calculations. [ 24 ] Curtarolo et al. designed a series of calculation procedures that can realize high-throughput calculations and finish the calculations of thermodynamic properties of more than 650 binary alloys and the calculations of electronic structures of 13000 inorganic structures in order to find radiation detection materials. [ 1 ] Steyawan et al. studied the electronic structure of 7439 compounds by means of highthroughput calculations. [ 25 ] Yang et al. found 28 topological insulators via high-throughput calculations. [ 26 ] Ceder’s group use this method to discover battery materials. [ 27 , 28 ] Besides the quantum mechanics-based methods, geometry-based highthroughput analysis has also been developed. Willems et al. proposed algorithms and tools to identify suitable crystalline porous materials. [ 29 ] High-throughput bond-valence calculations were adopted by Gao et al. to screen ionic conductors for solid electrolyte materials. [ 30 ]
The rapid development of high-throughput theoretical methods creates new modes for materials discovery, which make possible fast, computationally-derived materials design, screening and optimization.
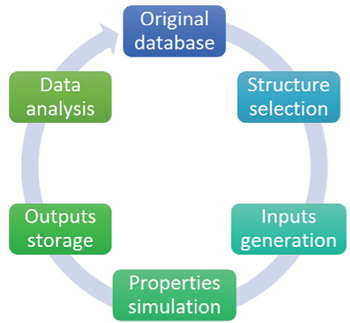
The recently developed high-throughput calculation methods are bases for new-materials discovery schemes. Although different calculations are needed for different types of materials, most high-throughput projects adopt DFT-based methods, since the quantum mechanical modeling provides perhaps the most accurate description of the energy and electronic structures. The keys to perform the high-throughput calculations are a series of automated flows for simulations of wide ranges of structures. The functions of the automatic simulation flows include structure selection, input generation, property simulation, output storage, data analysis, and database expansion, which together allow the tasks to be done automatically with minimal human intervention. [ 31 ] The overall relationship of the above steps is shown in Fig.
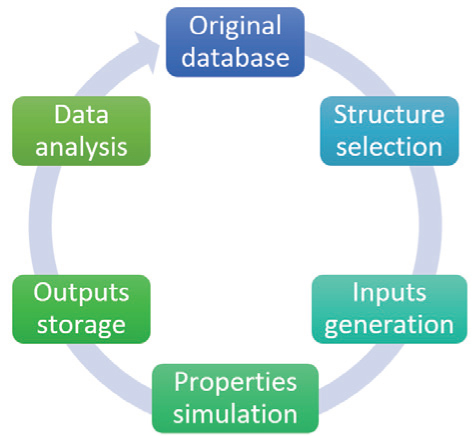 | Fig. 1. Data flow diagram in high-throughput computational materials design. |
Several groups have developed software to realize the automatic simulation flows for high-throughput calculations. Dolinsky et al. designed the PDB2PQR platform to facilitate the setup and execution of high-throughput continuum electrostatic calculations for biomolecular structures. [ 34 ] COSMO frag is a tool designed by Hornig et al. to evaluate the absorption, distribution, metabolism, and elimination parameters of drug candidates, and the pharmacokinetics are the main properties focused on. [ 35 ] Pronk et al. created a high-throughput simulation toolkit for biomolecules, named GROMACS, to carry out calculations supporting solvent models in parallel mode. [ 36 ] PYMATGEN is an open-source Python library for high-throughput materials analysis developed by Ceder’s group. [ 32 ] Jain et al. distribute another free code package, FIREWORKS, for defining, managing, and executing scientific computing workflows to perform high-throughput calculations at large computing centers. [ 37 ] Curtarolo et al. created a high-throughput framework AFLOWLIB to perform calculations for alloys. [ 1 ] These software packages are programmed for various types of materials screening. Most of them are integrated with simulation codes based on the density functional theory, and the main differences among them are the post-processing and analysis of the electronic structure data, since the properties of interest for each type of material differ widely. Thus, the specific quantities and criteria used for screening also differ. Besides the development of methods and software, efforts are also made to design more appropriate potentials for high-quality high-throughput calculations. For example, a comprehensive set of software and a transferable pseudopotential library has been optimized by Garrity et al. for high-throughput DFT calculations, and the accuracy has been validated for a variety of chemical environments, [ 38 ] which ensures the reliability of pseudopotential-based highthroughput DFT calculations in a wide range of materials. The rapid progress in both techniques and sciences makes the highthroughput theoretical design possible to apply in almost all material areas, including lithium battery materials. [ 27 , 28 ]
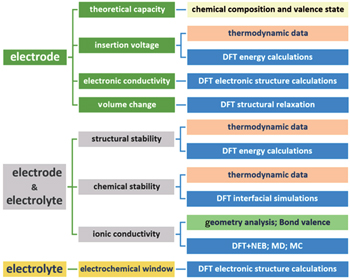
For the rational design of lithium battery materials, the set of candidate materials that can be screened or optimized at any given time is limited primarily by the range of properties that can be evaluated by the “ready” theoretical methods. The three fundamental components of lithium batteries are the positive electrode, the negative electrode and the electrolyte. For electrode materials, the capacity, insertion voltage, electrical conductivity, lithium diffusion coefficient, and volume variation during charge and discharge are the main properties of interest. For electrolyte materials, ionic conductivity, electrochemical window, structural stability and chemical stability between the electrode and the electrolyte are the most important factors. A variety of calculation methods with different bases and accuracies, such as thermodynamics, bond-valence, first principles calculation, molecular dynamics, Monte Carlo simulations, etc., can be used to calculate the properties mentioned above. Figure
 | Fig. 2. The properties (middle column) of electrode and electrolyte materials that can be calculated and the methods (right column) used to simulate them. |
For electrode materials in lithium batteries, the theoretical capacity is closely associated with the numbers of electrons and lithium ions that can be transferred in the electrode. For a certain electrode material, the capacity can be calculated by [ 39 ]
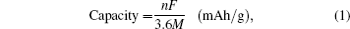
The average intercalation potential of electrode materials is closely associated with the Gibbs free energy of the electrode reaction, so the average intercalation voltage can be calculated as
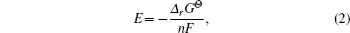
The precise and quantitative calculation of electrical conductivity is not an easy task. The compromising way is to make a qualitative estimate according to the electronic structure obtained from DFT calculations. The volume change of the electrode materials during charge and discharge cycles can be obtained with reference to the relaxed cell volume by using DFT calculations for structures with different lithium concentrations.
Structural stability and chemical stability are properties necessarily considered for both electrode and electrolyte materials. For stabilities, thermodynamic data can be used for preliminary estimation of the tendency toward presumed reactions, e.g., phase transition, decomposition, the reaction between electrode and electrolyte, etc. Accurate energy calculations based on quantum mechanical modeling can provide a theoretical prediction of stability, from the thermodynamic point of view.
Ionic transport is one of the most important factors that greatly affect battery performance. In solid or liquid, lithium ions can be transferred by jumping to vacant or interstitial sites, forming the macro transport. During diffusion, the migrating lithium ion is hindered by a crystal field formed by other ions, and the effect of this hindrance is evaluated by the activation energy. The approaches to understanding ionic migration in solids start with geometric analysis of the net channels in a specific crystalline structure using the model of excluded volume and Voronoi–Dirichlet partitioning. [ 41 , 42 ] To consider the interactions among atoms, bond-valence and force-based bond-valence theory are introduced to find sites accessible by mobile ions and roughly estimate the activation energies. [ 43 , 44 ] More reliable calculations of energy barriers can be obtained from the transition-state method, like the nudged elastic band (NEB) method, or the molecular dynamics method based on density functional theory (DFT). [ 45 – 47 ] Monto Carlo simulations can provide information on ionic conductivities, even for phases with inhomogeneous lithium distribution.
For electrolytes, the electrochemical window is another important screening criterion besides the ionic conductivity. The electrochemical window represents the voltage range within which the electrolyte is stable, and it is believed that the width of the window is closely related with the band gap of the electrolyte, [ 48 ] which can be obtained by DFT electronic structure calculations.
Among the methods mentioned above, the estimation based on thermodynamic data relies on the experimental results of existing materials, which pose certain limitations for the discovery of new materials. The methods based on geometric analysis or empirical potential energy function are fast techniques, although with limited accuracy. The calculations based on DFT provide perhaps the most accurate description of structures, energies and electronic structures but suffer from high computation cost, which limits their efficiency. Because of the distinctive features of each type of method, combinations of the above approaches at different stages may be a more practical scheme for discovering lithium battery materials. The common feature for these methods is that the input data of the calculations depend only on crystalline structure or bonding information, which provides possibilities of automatically generating initial data for simulations. Thus, highthroughput calculations based on the above methods can be realized for lithium battery materials.
By using the calculation methods for lithium battery materials described in the last section, the theoretical design scheme for new electrodes and electrolytes can start from rational high-throughput screening. The screening criteria and procedures are presented in Ref. [ 49 ] and illustrated here as Fig.
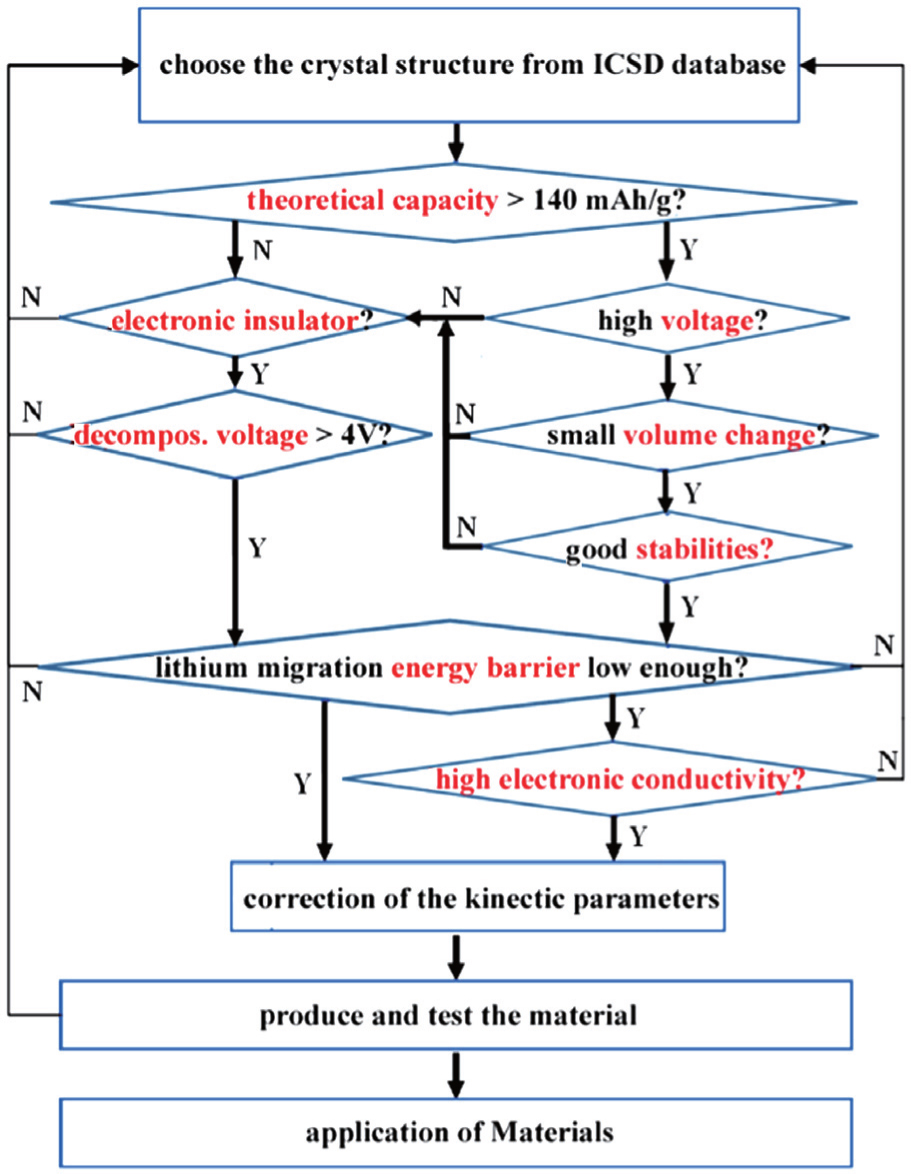 | Fig. 3. Procedures of screening electrode materials and solid electrolytes using high-throughput calculation method. [ 49 ] |
Before performing time-consuming ab initio calculations, a preliminary screening based on the high-throughput thermodynamic calculations is truly necessary. According to Eqs. (
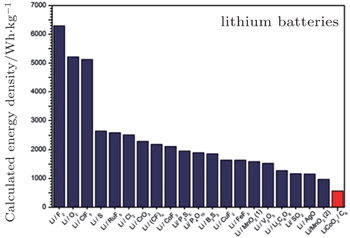
 | Fig. 4. Calculated energy densities of different lithium battery systems according to the Nernst equation. [ 39 ] |
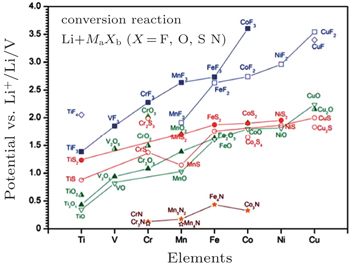
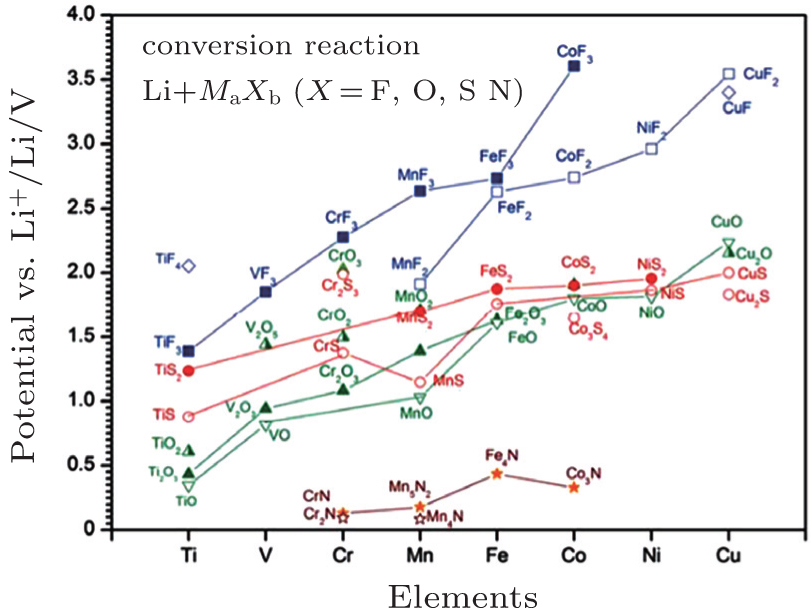 | Fig. 5. Calculated thermodynamic equilibrium voltage of conversion reactions between binary transition metal compounds and lithium. [ 39 ] |
In the research of lithium battery materials, Ceder’s research group was first to launch high-throughput ab initio calculations to discover high-capacity cathodes. [ 27 , 28 ] They carry out the high-throughput ab initio analysis for both phosphates [ 27 ] and polyanions. [ 28 ] In these studies, substitution for host elements in the original structure is the main approach to creating new structures from the crystal database. The calculations involve the capacity, voltage, specific energy, energy density and thermal stability of potential materials and extend to thousands of compounds. For phosphates, three categories are considered in the calculations, including orthophosphates with an O/P ratio of 4, oxyphosphates with an O/P ratio larger than 4, and condensed phosphates with an O/P ratio between 2.5 and 5. [ 27 ] The thermodynamic stability of phosphates is analyzed at zero K and zero pressure according to the DFT energy calculations, and the capacity limits for three types of phosphates are given. In Fig.
 | Fig. 6. Mean voltage in phosphates versus maximum gravimetric capacity achievable obtained by high-throughput DFT calculations. The upper voltage considered to be a threshold safe against decomposition of the electrolyte is indicated by the red dashed line. [ 27 ] |
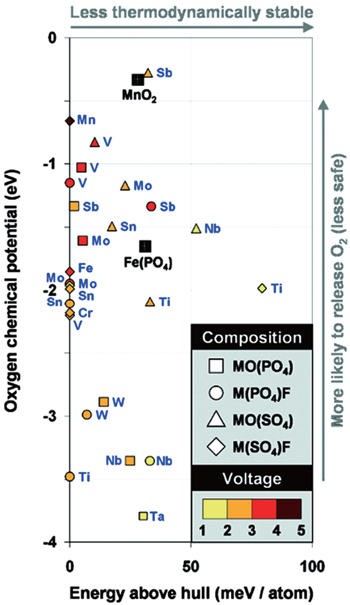
The mineral tavorite is another type of structure interesting Ceder’s group. Mueller et al. calculated average voltages of insertion of one and two lithium ions per redox-active metal and evaluated the stability of each candidate against a database of nearly 100000 compounds. [ 50 ] Figure
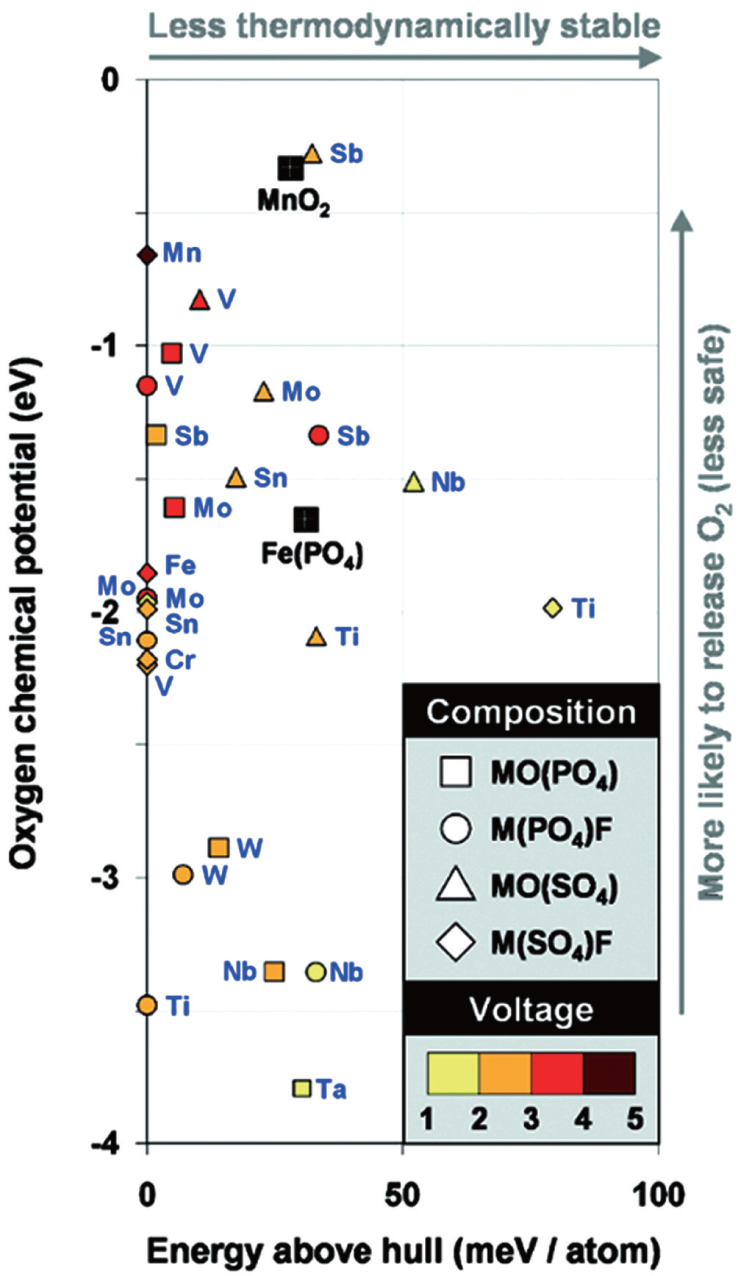 | Fig. 7. Energy relative to the thermodynamic hull and oxygen chemical potential for fully delithiated tavorite-structured cathode materials by high-throughput DFT calculations. [ 50 ] |
Besides high capacity, long life is another important target for lithium battery design. However, a simple physical description of the cycle life of lithium batteries is still not easy to grasp because the degradation rate during charge/discharge cycles is influenced by many factors. It is already widely accepted that cycle life is related to the volume change of the electrode during lithiation and delithiation, because too much volume variation induces stress inside the electrodes and microcracks may form, weakening performance. To accelerate the discovery of cathodes with longer cycle life, Nishijima et al. take the relative volume change of a compound between fully lithiated and delithiated conditions as a criterion to screen materials with LiFePO 4 structure. [ 51 ] Substituting solute elements on the different cation sites of the LiFePO 4 structure, high-throughput DFT simulations with around 2000 calculations are carried out in a wide chemical composition space. Both the relative volume change and the planar mismatch of (100) planes between the lithiation and delithiation phases are obtained from the calculations. According to the theoretical screening results, Nishijima et al. synthesized targeted materials Li(Fe 1− x Zr x )(P 1−2 x Si 2 x )O 4 and test their performance in a full cell configuration. A prolonged cycle life was confirmed by experimental measurements, and the estimated life is up to 25000 cycles for 70% capacity retention at x = 0.050. [ 51 ] The combination of high-throughput DFT calculations and targeted experimental synthesis and tests demonstrate the effectiveness of high-throughput theoretical design of cathode materials.
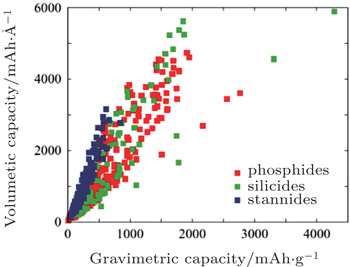
Compared to the diversity of cathode materials, the range of candidate anode materials is much narrower. Graphitic carbon based materials are the dominant anodes for the current generation of commercial lithium-ion batteries. [ 52 ] However, to pursue more compact, long-lasting batteries, finding new anode materials with better performance is necessary. Kirklin et al. proposed to search for new candidates in the class of alloying-reaction anodes, including silicon, phosphorus and tin. [ 53 ] By performing high-throughput DFT calculations on 515 thermodynamically stable lithiation reactions, the cell potential, volume expansion and capacity for all transition metal silicides, phosphides and stannides found in the ICSD database are evaluated. Figure
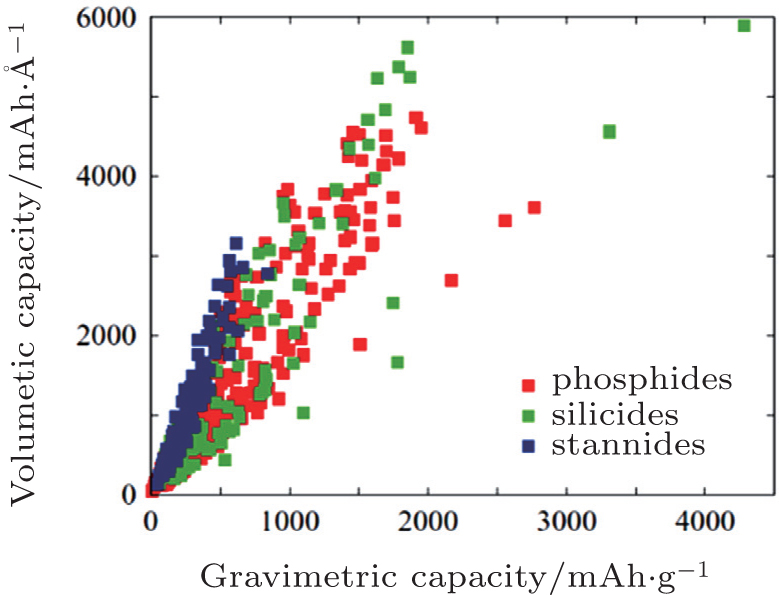 | Fig. 8. High-throughput DFT calculated gravimetric and volumetric capacities for all transition metal-stannide, -phosphide and -silicide lithiation reactions. [ 53 ] |
In order to solve the safety issue of lithium batteries, development of solid state electrolytes has become very urgent. Solid state electrolytes have at least three advantages over today’s liquid electrolytes. First, the safety of the battery is expected to be better because of the stability and non-flammability of inorganic solid electrolytes. Second, the energy density of the battery may be greater because of the higher voltage that is possible. Third, the choice of cathode material will be more flexible, because the lithium content will not be a requirement since lithium metal is potentially used as an anode. One of the challenges in developing solid state lithium batteries is to discover solid state electrolyte materials with good ionic conductivity. Some inorganic electrolytes have already been developed, including NASICON-type (LiZr 2 (PO 4 ) 3 ), LISICONtype (Li 4 ZnO 3 -Li 4 GeO 4 ), garnet-type (Li 7 La 3 Zr 2 O 12 ), and perovskites (Li 0.5 La 0.5 TiO 3 ). Recently, researchers from Japan developed a new electrolyte material, Li 10 GeP 2 S 12 , with ionic conductivity up to 12 mS/cm at room temperature, [ 54 ] a value near to the order of magnitude in liquid electrolytes. Although these materials show relatively high ionic conductivities, the practical application of them in all-solid-state lithium batteries is hindered by other shortcomings such as the small electrochemical window, unstable interfacial stability with electrode materials, etc. To select candidate electrolyte materials from a wide range of compounds, high ionic conductivity is the most important requirement. However, basing precise simulations of lithium migration energy barriers and lithium-ionic conductivity on DFT+NEB or molecular dynamics is computationally expensive, limiting the efficiency of simulations. One alternative is to screen candidates by a less accurate but faster method before undertaking detailed studies with DFT methods. The bond-valence method or the bond-valence based force-field method would be very suitable for the initial screening. [ 55 ] By linking the bond-valence mismatch to the absolute energy scale, using a general Morsetype interaction potential, both the ion migration paths and energy barriers can be extracted from the energy landscape simulated by this energy-scaled bond-valence method. [ 55 ] Gao et al. carried out a high-throughput bond-valence calculation on more than 1000 structures from the ICSD database, and obtained the lithium conduction pathways for them. [ 30 ] The high-throughput bond-valence based force-field method can provide further information on lithium migration energy barriers. Figure
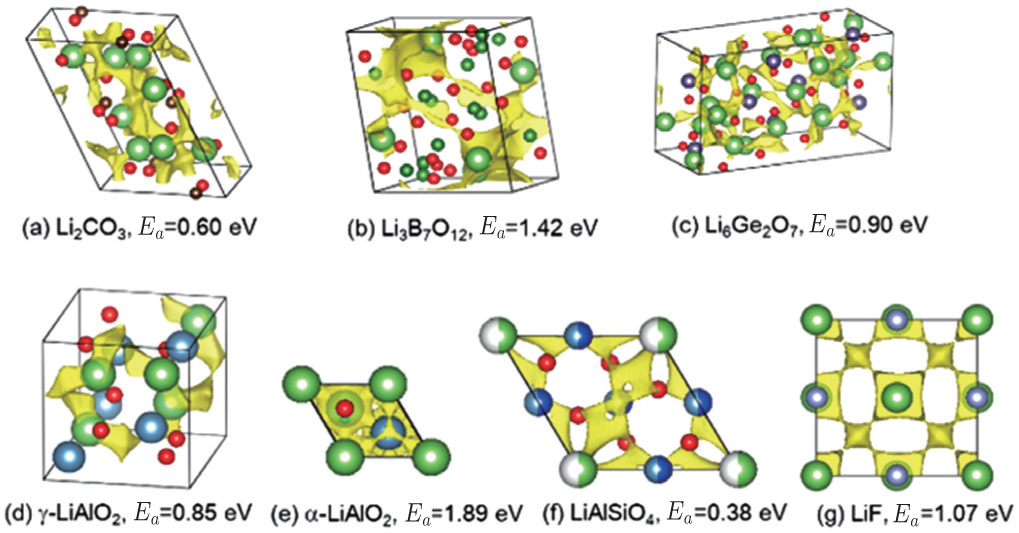 | Fig. 9. The pathways and energy barriers of Li + migration for seven compounds calculated by the bond-valence based force-field method. [ 56 ] |
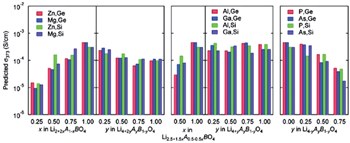
The preliminary screening can be completed for solid state electrolytes by the bond-valence method, but more accurate calculations based on DFT methods are necessary to perform a more detailed screening and optimization on the selected structures. Fujimura et al. carried out systematic sets of first-principles molecular dynamics simulations on LISICONtype electrolytes to optimize the ionic conductivities in this structure. [ 57 ] They performed an extensive set of calculations on the Li 8− c A a B b O 4 system with A = Zn, Mg, Al, Ga, P, or As, and B = Si or Ge. By fitting the Arrhenius plots of calculated Li-ion diffusion coefficients, the activation energies are obtained for each composition. Besides simulating kinetic behavior, they also predicted the conductivity of each composition at low temperature (373 K) through a machinelearning technique by the combination of theoretical and experimental datasets. Figure
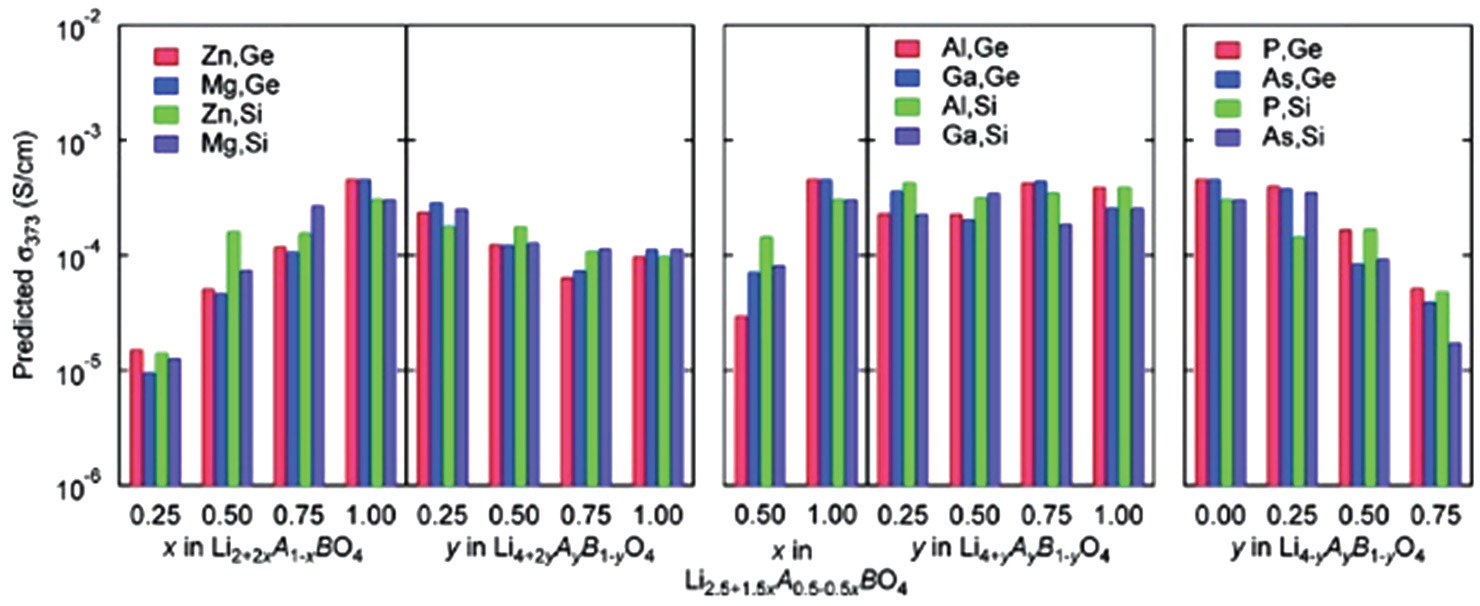 | Fig. 10. Predicted ionic conductivities at 373 K for 72 compositions of the LISICONs Li 8− c A a B b O 4 system. [ 57 ] |
The liquid electrolytes currently used in lithium-ion batteries often consist of one or more lithium salts dissolved in an aprotic solvent and at least one additive, which may serve to enhance electrode stability. [ 58 ] Unlike electrode materials consisting mostly of inorganic crystals, the additives mainly include organic carbonates and their derivatives. To discover electrolyte additives for lithium-ion batteries, Halls and Tasaki created an additive library of 7381 structures, containing highthroughput quantum chemistry calculations of the optimized geometries, HOMO and LUMO energies, vertical ionization potentials, electron affinities and chemical hardness of these structures. [ 58 ] The structures are virtually produced based on ethylene carbonate, and the calculations are carried out using the PM3 semi-empirical method. This additives library is an effective tool to accelerate the screening and optimization of electrolyte additives through the whole chemical design space.
High-throughput theoretical design plays an important role in the discovery of new materials, and it can be expected to be increasingly effective in looking for and designing new lithium battery materials in the future. Based on the structures in the ICSD database and their derivatives, promising candidates for the next generation of electrodes and solid state electrolytes can be identified by choosing reasonable search criteria. The possible existence of new structures and their properties can be predicted in a high-throughput way. Thus, the process from the discovery of a material to its application can be shortened, and the development of new battery materials, even new battery systems, can be supported. Moreover, tons of data will be created during these high-throughput calculations, providing rich resources for systematically mining and analyzing the relationship between structure and properties. The structure–properties relationship will further guide us in optimizing and designing materials with better performance. In the near future, high-throughput theoretical simulations combined with high-throughput experiments are likely to be applied more and more in the discovery of new lithium battery materials.
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 | |
| 22 | |
| 23 | |
| 24 | |
| 25 | |
| 26 | |
| 27 | |
| 28 | |
| 29 | |
| 30 | |
| 31 | |
| 32 | |
| 33 | |
| 34 | |
| 35 | |
| 36 | |
| 37 | |
| 38 | |
| 39 | |
| 40 | |
| 41 | |
| 42 | |
| 43 | |
| 44 | |
| 45 | |
| 46 | |
| 47 | |
| 48 | |
| 49 | |
| 50 | |
| 51 | |
| 52 | |
| 53 | |
| 54 | |
| 55 | |
| 56 | |
| 57 | |
| 58 |