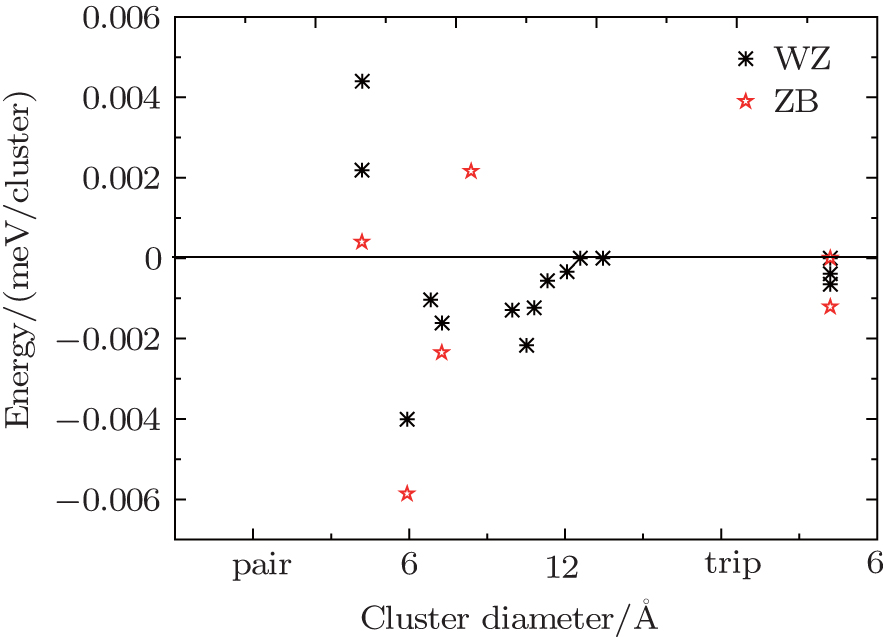
 Table 1. ECIs of the wurtzite and zinc-blende Cd 1– x Zn x S alloys. . | Phase | Cluster | Coordinates | Size/Å | Multiplicity | ECI |
|---|
| WZ | J (0,1) | | | 1 | 0.06875 | | J (1,1) | (0.333,0.667,0.003) | | 2 | 0.00214 | | J (2,1) | (0.667,0.333,0.503) (0.333,–0.333,1.003) | 4.184 | 6 | 0.00219 | | J (2,2) | (0.667,0.333,0.503) (–0.333,–0.667,0.503) | 4.196 | 6 | 0.00440 | | J (2,3) | (0.333,0.667,0.003) (–0.333,–0.667,–0.497) | 5.925 | 6 | –0.00401 | | J (2,4) | (0.667,0.333,0.503) (-0.333,-0.667,-0.497) | 6.822 | 2 | –0.00105 | | J (2,5) | (0.333,0.667,0.003) (1.667,2.333,–0.497) | 7.268 | 6 | -0.00161 | | J (2,6) | (0.667,0.333,0.503) (–0.333,-1.667,1.503) | 9.968 | 6 | –0.00129 | | J (2,7) | (0.333,0.667,0.003) (0.667,1.333,–1.497) | 10.516 | 6 | –0.00217 | | J (2,8) | (0.333,0.667,0.003) (–1.667,–1.333,–0.997) | 10.815 | 12 | -0.00123 | | J (2,9) | (0.333,0.667,0.003) (–0.333,–1.667,–1.497) | 11.322 | 6 | –0.00056 | | J (2,10) | (0.667,0.333,0.503) (-0.667,-1.333,2.003) | 12.075 | 12 | –0.00033 | | J (2,11) | (0.333,0.667,0.003) (–2.333,-2.667,0.503) | 12.584 | 6 | 0.00000 | | J (2,12) | (0.333,0.667,0.003) (–1.333,–1.667,–1.497) | 13.454 | 12 | 0.00000 | | J (3,1) | (0.667,0.333,0.503) (0.333,–0.333,1.003) (–0.333,-0.667,0.503) | 4.196 | 6 | -0.00065 | | J (3,2) | (0.667,0.333,0.503) (0.333,–0.333,0.003) (–0.333,-0.667,0.503) | 4.196 | 6 | –0.00038 | | J (3,3) | (0.667,0.333,0.503) (–0.333,0.333,0.503) (–0.333,–0.667,0.503) | 4.196 | 2 | 0.00000 | | ZB | J (0,1) | | | 1 | 0.12673 | | J (1,1) | (1.00,1.00,1.00) | | 4 | 0.00424 | | J (2,1) | (1.00,0.50,0.50) (1.50,0.50,0.00) | 4.191 | 24 | 0.00040 | | J (2,2) | (0.50,0.50,1.00) (1.50,0.50,1.00) | 5.927 | 12 | –0.00586 | | J (2,3) | (0.50,0.50,1.00) (1.50,1.00,1.50) | 7.259 | 48 | –0.00235 | | J (2,4) | (0.50,0.50,1.00) (1.50,0.50,0.00) | 8.382 | 24 | 0.00216 | | J (3,1) | (1.00,0.50,0.50) (1.50,0.50,0.00) (1.00,0.00,0.00) | 4.191 | 16 | -0.00122 | | J (3,2) | (1.00,0.50,0.50) (0.50,0.50,0.00) (1.00,0.00,0.00) | 4.191 | 16 | 0.00000 |
| Table 1. ECIs of the wurtzite and zinc-blende Cd 1– x Zn x S alloys. . |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 , Tang Fu-Ling 1, ‡,
, Tang Fu-Ling 1, ‡,