



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Recent technical advancements enabled atomic resolution CryoEM
[Li Xueming †,  ]
]
]
|
|


† Corresponding author. E-mail:
Project supported by Tsinghua–Peking Joint Center for Life Sciences, China.
With recent breakthroughs in camera and image processing technologies single-particle electron cryo-microscopy (CryoEM) has suddenly gained the attention of structural biologists as a powerful tool able to solve the atomic structures of biological complexes and assemblies. Compared with x-ray crystallography, CryoEM can be applied to partially flexible structures in solution and without the necessity of crystallization, which is especially important for large complexes and assemblies. This review briefly explains several key bottlenecks for atomic resolution CryoEM, and describes the corresponding solutions for these bottlenecks based on the recent technical advancements. The review also aims to provide an overview about the technical differences between its applications in biology and those in material science.
The transmission electron microscope (TEM) was invented in the 1930s with the discovery of the wave-particle duality of electrons. Due to the controllable and short wavelength of the high-energy electron beam, usually much less than 0.1 Å, the resolution of TEM is practically unlimited by the optical diffraction limit. Hence, TEM was initially developed as a tool for direct observation of the atomic structure of materials. After its invention, the atomic resolution of 1–2 Å has become widely achievable in material science. TEM was also introduced into structural biology to study the structure of cells and bio-macromolecules. Due to the requirement for a hydrated environment, the biological sample must be frozen in vitrified ice to avoid dehydration by the high vacuum of the electron microscope. The corresponding TEM technique is called electron cryo-microscopy (CryoEM). Considering the capability of TEM, CryoEM was also expected to have the same resolution as its counterpart in material science. However, it is not a simple matter.
The goal of CryoEM is to reconstruct the three-dimensional (3D) structure of a biological sample under its native condition, which helps biologists to understand the dynamic processes in life. Based on the central section theorem, the 3D reconstruction is calculated by merging a series of two-dimensional (2D) projection images from different but identical molecules, termed single-particle CryoEM (Fig.
Due to the high energy of the electron beam (typically 80– 300 keV) and the strong interaction between electrons and the biological material, the biological sample must be imaged with a very low dose, typically less than ∼ 20 e/Å 2 , before being seriously damaged by the electron beam. Such a low dose leads to low contrast and a low signal-to-noise ratio (SNR) of the CryoEM image, accordingly, the difficulties in determining the orientations of projection images, causing misalignment. The misalignment has a major effect on the resolution of the 3D reconstruction. To compensate for the problems caused by a low dose, CryoEM has to work with very different imaging conditions from those in material science, such as micrometer defocus, to improve the image contrast and low magnification to obtain a sufficient amount of projections. Such conditions reduce the performance of TEM at atomic resolution range. Compared with the TEM application in material science, the differences in both image processing technologies and TEM operating conditions make atomic resolution a difficult task for CryoEM. Until the latest decade, successful examples reaching near atomic resolution of ∼ 3–4 Å by CryoEM were still limited to large viruses with icosahedral symmetry. [ 4 – 6 ] For small and low-symmetry molecules between ∼ 100 kDa and ∼ 1 MDa, the resolution of CryoEM is mostly in nanometer range or worse. Only the shape of certain secondary structure, such as α -helix, can be resolved. To improve the resolution, new techniques have been developed to overcome issues caused by low dose and un-optimized TEM conditions. This review will briefly explain several key bottlenecks limiting the resolution and corresponding solutions that have caused a shift toward atomic-level resolution of single-particle CryoEM.
 | Fig. 1. Basic procedure of single-particle electron cryo-microscopy (CryoEM). The purified protein complex or assembly is frozen in a thin layer of vitrified ice (a) with presumably random orientation. The plus signs denote the positive static charges on the surface of ice illuminated by a high-energy electron beam, which is one of the reasons for image motion. The sample is then imaged to obtain different views (boxed particles in the panel (b) of the complex or assembly). Due to the low dose and corresponding low signal-to-noise-ratio (SNR) of the image, only the overall shape of particles is directly visible. After a 3D alignment procedure, many such particles are orientated and merged in 3D space to reconstruct a 3D density map (c). The side-chain details become visible in the map. |
With improvements in both hardware and software, such as parallel electron source, stable electron optical system and decent image processing technology, the resolution of singleparticle CryoEM was gradually pushed to nanometer level in the past decade. However, until three years ago, except large high-symmetric viruses, it is still a challenge for molecules smaller than 1 MDa to achieve the resolution at atomic level. The following factors have been thought of as the key bottlenecks limiting the higher resolution.
To obtain the structural information of a biological sample, the image should be first taken and digitalized by a camera. Although the electron cryo-microscope has to be operated under a micrometer defocus condition which is not optimal for performance, most modern TEMs equipped with a field emission gun are still able to deliver structural information better than 2 Å resolution to the camera plane. Therefore, camera performance is critical for the obtainable resolution of the next 3D reconstruction step.
There are at least three types of image recording media or cameras that are used for CryoEM, i.e., film, charge-coupled device (CCD) camera and complementary metal-oxide semiconductor (CMOS) camera. There is also another image recording medium, the electronic image plate, which is mostly used to record electron diffraction and is seldom used in CryoEM and will not be discussed here. Both film and cameras first convert the incident electrons to detectable light signal by a phosphor scintillator layer, and then record the light intensity by either a scanner or an electronic detector. During the conversion and recoding step, the input signal might be attenuated and extra noise might be added. [ 7 – 9 ] To characterize the performance of an image recording system, the detective quantum efficiency (DQE) and modulation transfer function (MTF) have been widely adopted. Because of combining the influences from most of the performance-related factors, including MTF (discussed later), DQE is thought to be a comprehensive indictor of camera performance. DQE describes the squared ratio of the output and the input SNR at different spatial frequencies in Fourier space. [ 10 , 11 ] The DQE of traditional cameras usually decays rapidly as the spatial frequency increases, typically from ∼ 0.4 at low frequency to ∼ 0.2 or worse at high frequency. [ 11 ] For a digital image, the maximum spatial frequency in the Fourier transform of the image is half of the reciprocal of the image pixel size, termed Nyquist frequency. In material science, the sample is usually imaged at high magnification, which corresponds to an over-sampled small pixel size and wide spatial frequency range. All use-ful signals in Fourier transform of the image, typically within ∼ 1 Å − 1 range, are located in the low-frequency region of the camera, where the camera has the best performance. Together with the high dose and corresponding high SNR, the decay of ∼ 0.4 is a very subtle influence on the image analysis. However, for a biological sample, much lower magnification has to be used to cover sufficient imaging area in order to increase the efficiency of data collection or cover a large enough cellular area within a single image. The low magnification leads to a large pixel size and, accordingly, a spatial frequency range covering most of the spatial frequency space of the camera. Consequently, camera performance at high frequency is very critical.
The low-frequency signal contributes to image contrast which is important to recognize the biological sample embedded in vitrified ice and to guide the 3D alignment. The highfrequency signal contains high-resolution details, for example, the side chain of the amino acid and ligand bound to the protein. Under the low-dose imaging condition, the input SNR of a biological sample is often less than 0.1. The additional signal decay and noise from the camera, especially at high resolution, smear out the high-resolution structural information to undetectable level. For this reason, the low performance of traditional cameras already makes the atomic resolution impossible before the subsequent image-processing step.
A biological sample must be stabilized at the angstrom level to obtain nanometer or atomic resolution. There are at least two types of sources of instability. The first is mechanical motion or vibration. Unstable specimen stage, temperature gradient between the biological sample holder cooled by liquid-nitrogen and the microscope at room temperature, as well as environmental vibrations cause sample drift. The second source is the so-called beam-induced motion. [ 12 ] When the high-energy electron beam hits the biological sample and stimulates the escape of secondary electrons, the surface of the insulated biological sample will be charged (Fig.
Due to the low SNR and the requirement for orientation coverage in single-particle CryoEM, thousands or even millions of images are merged to obtain the 3D reconstruction of a target sample. Although these images are taken from different molecules, it is assumed that all molecules have the same conformation. The heterogeneous or flexible region in molecules will be averaged out and will disappear in the final reconstruction. Even more seriously, the heterogeneity can cause the failure in determining 3D orientation parameters against the same 3D reference, and lead to overall low resolution or even wrong results.
Heterogeneity in conformation or composition is usually a natural property of biological macromolecules and coherent with its biological function. For the x-ray crystallographic approach, heterogeneity must be carefully removed through a series of biochemistry modifications, such as mutations and truncations, before a successful crystallization. Without the need for crystallization, CryoEM has a slightly higher tolerance for sample heterogeneity, if the heterogeneity is limited to a small region of a large molecule. However, in practice the requirement for overall homogeneity is almost as critical as that for the crystallographic approach. Stabilizing and isolating homogenous molecules are always necessary to reach atomic resolution by single-particle CryoEM.
Several technical breakthroughs [ 16 – 18 ] in 2013 have provided solutions for the key bottlenecks mentioned above and brought CryoEM into a new era of atomic resolution. Immediately, the applications of these new techniques have exhibited the power of CryoEM by solving a series of atomic resolution structures. [ 19 – 22 ] The following sections will briefly discuss these new technical achievements and how they overcome the key bottlenecks.
In the past five years, a new type of camera, named direct detection detector (DDD) camera, was developed and commercially available. The DDD camera has a much higher DQE than traditional cameras, and provides improved contrast and SNR at high resolution. [ 16 , 23 – 26 ] Such improvements make the structure determination of small molecules and atomic resolution more reachable than ever. Currently, there are three commercially available DDD cameras, i.e., the DE series camera (Direct Electron Company, San Diego, CA, USA), Falcon camera (FEI Company, Hillsboro, OR, USA), and K2 Summit camera (Gatan Company, Pleasanton, CA, USA). The first two cameras linearly accumulate the signal stimulated by the incident electron (referred to as DDD linear cameras below). In particular, the K2 camera works in a different mode, named electron counting, which recognizes each primary electron and digitally counts the electron inputs (referred to as DDD counting camera below). [ 16 ] The electron counting technology further improves the DQE on top of the DDD linear camera, and makes the counting camera closer to an ideal pixelated camera.
The point spread effect from the signal spreading around the incident electron is one of the major causes of low camera performance. It occurs for each incident primary electron and is mathematically indicated by convoluting a point spread function (PSF) to each point on the image. [ 10 , 11 ] Accordingly, the Fourier amplitude, calculated by the Fourier transform of the image, is modulated by the Fourier transform of the PSF, named the modulation transfer function (MTF). The MTF usually has a similar shape to the Gauss function and decays from 1 at zero frequency to nearly 0 at high frequency.
For a long time, the digital camera used for the electron microscope could not detect electrons directly, partially owing to the radiation hardness of the semiconductor detector. A scintillator was then used to convert incident electrons to detectable photons, and coupled by either lenses or optic fibers to transmit photons to the camera detector. The procedure from electrons to detectable photon signals produces a wide PSF, correspondingly, an MTF with rapid decay, which attenuates the signal at high frequency. To improve the PSF and MTF, efforts were invested on thinning and even removing the scintillator to obtain sharper PSF. [ 23 , 25 ] Finally, DDD linear cameras without scintillator were developed. To further reduce the influences of back-scattered electrons in the detector, the bulk support for the detector chip was removed, referred to as back-thinned DDD linear camera, to maximize the camera performance. [ 27 ]
In addition to the significantly improved MTF, the direct detection technology also eliminates readout noises associated with the scintillator, fiber optics, or lenses. Owing to the combined improvements in MTF and camera noise, the DQE of a DDD linear camera is superior to that of traditional cameras. [ 23 , 25 , 27 ]
The structural information of a biological sample is carried by the electron wave and presented as the number of electrons in different spatial positions, proportional to the probability density function of the beam. Therefore, the ideal image recording is to count the number of electrons striking each pixel. [ 24 ] For this purpose, the first electron-counting camera (K2 Summit TM , Gatan) was developed using a different mechanism from those of traditional and DDD linear cameras. The K2 camera has an intrinsic high frame rate, i.e., 400 frames/sec, enabling each primary electron to be individually recorded. [ 16 , 28 ] Assisted by high-speed computing, each frame is immediately analyzed to centroid and the incident primary electrons counted within this frame (Fig.
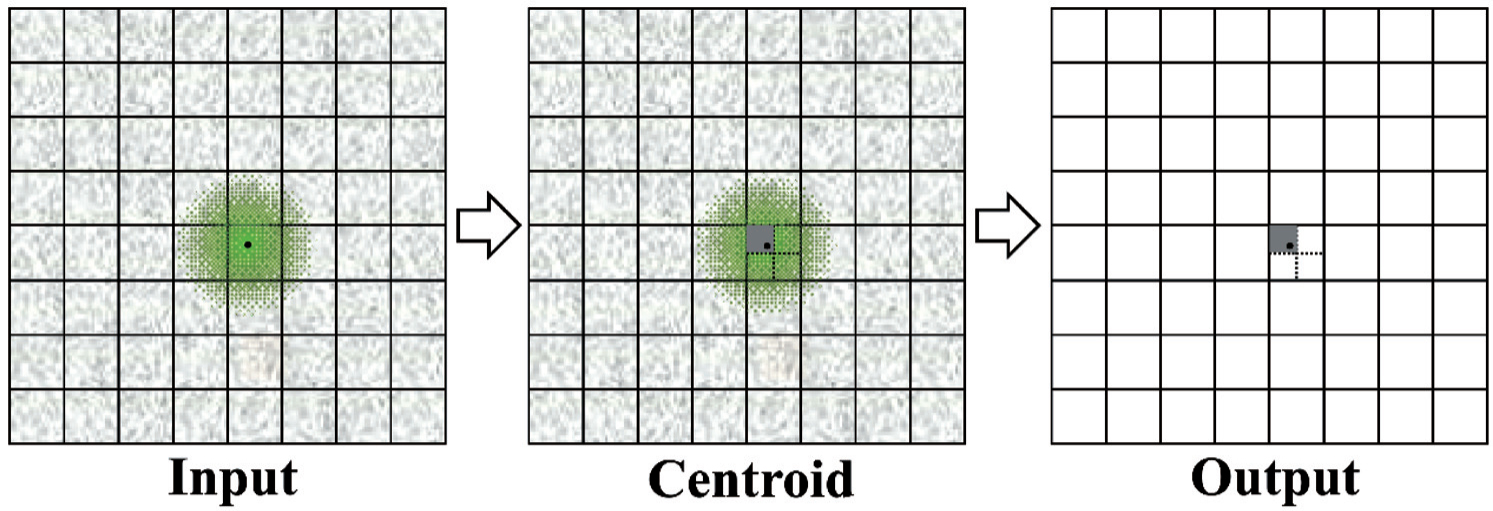 | Fig. 2. Diagram of counting and centroiding with half-pixel precision. The lattice of squares denotes the pixel matrix of a counting camera, and the noisy background represents the readout noise. The small black dot denotes an incident primary electron which stimulates signals (green spot) around it. The peak position of the green spot is centroided with half-pixel precision, and then its intensity is digitalized and counted as one. Consequently, the point spread effect and readout noise are eliminated from the final output image. |
Compared with the DDD linear camera, counting has at least three advantages. [ 28 ] First, the point spread effect caused by electron striking is totally removed by the counting procedure. Second, the camera is almost free of noise. During counting, only the number of electrons is digitally counted, and all the background noise that appears in the intrinsic frame is discarded. For the DDD linear camera, the remaining pointspread signal together with all background readout noise and Landau noises (from the statistical energy fluctuation when each primary electron strikes the camera) are accumulated. Third, the intensity variation of electron striking is eliminated. The signal stimulated by electron striking is usually different for each electron and varies with incident position. By counting, the intensity of each electron is digitalized to one, hence, the detected intensity differences among electrons are eliminated, while DDD linear camera and traditional cameras simply accumulate all the electron signals and cannot correct this intensity variation. The intensity variation usually contributes to low-frequency noise of the image. [ 29 ] Therefore, the counting camera has less low-frequency noise than the traditional cameras.
Considering the above advantages, the DQE of the counting camera is significantly improved, that is, much stronger signal at both low- and high-frequency range can be recorded than the accumulation mode of DDD linear cameras (Fig.
 | Fig. 3. (a) DQE of three cameras. [ 16 ] Gatan UltranScan Camera (black) is a traditional camera with a scintillator. The accumulation mode (blue) and counting mode (red) of the K2 camera represent the DDD linear camera and counting camera, respectively. Note that centroiding with half-pixel precision doubles the camera size. (b) Rotational average of the power spectrum of two images of Pt/Ir film, respectively taken with the linear accumulation mode (blue) and counting mode (red) of the K2 camera. [ 28 ] The inset figure shows the spectrum SNR (SSNR) calculated from the two curves in the main figure, where the higher red curve (counting mode) confirms the DQE observed in panel (a). |
 | Fig. 4. (a) An image of a frozen hydrated archaeal 20S proteasome sample taken on a TVIPS F816 traditional CMOS camera with 1.6 μm defocus and 200 kV accelerating voltage, and (b) its power spectrum. Panels (c) and (d) are similar to panels (a) and (b), respectively, but were taken with a K2 summit counting camera with 1.1 μm defocus and 300 kV accelerating voltage. |
However, the counting camera must be operated with a relatively weak beam, i.e., using a low dose rate. [ 28 ] When more than one electron strikes the same position, the counting camera is not able to distinguish them and just simply counts them as one electron. Consequently, the other electrons are undercounted. This effect is called coincident loss, and must be carefully considered during the application. For the K2 counting camera with an intrinsic frame rate of 400 frames/sec, the dose rate of ∼ 10 e/pixel/sec or lower is recommended. The disadvantage of the low dose rate is that a prolonged exposure time must be used, typically 5–20 s. Therefore, the image will be more sensitive to specimen instability than traditional and DDD linear cameras. Fortunately, the motion during a long exposure can be compensated by motion correction technology (discussed below).
A benefit of the DDD camera is its high output frame rate. All commercially available DDD cameras can output up to 40 frames per second, called dose fractionation or movie mode. That is, within one exposure, the DDD camera can sequentially take multiple images (frames) rather than a single image. This feature brings the possibilities of analyzing and correcting imperfections in an image, which was previously impossible.
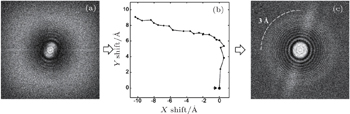
Motion correction was the first technique that benefited from dose fractionation. As mentioned in the previous section, motion from the specimen instability blurs an image, especially high-frequency (high-resolution) signals (Fig.
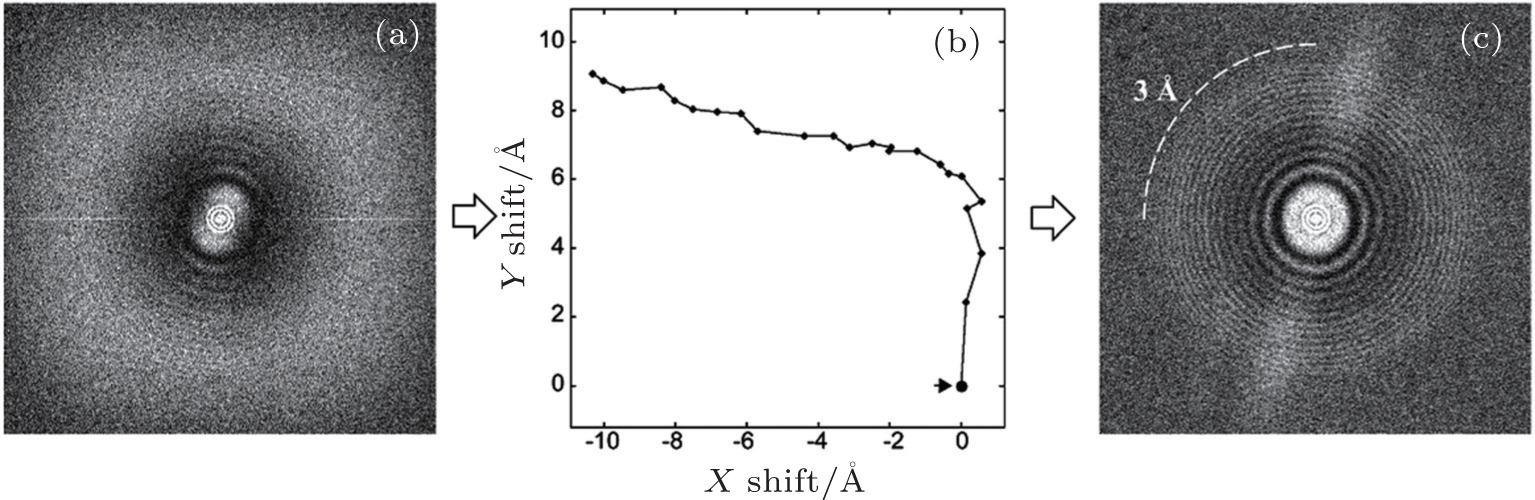 | Fig. 5. Thon ring before and after motion correction. [ 16 ] (a) The power spectrum of an image with motion. The Thon ring is smeared by the motion. (b) The trace of motion in panel (a), which is measured using the redundant approach. (c) The power spectrum after motion correction. The smeared Thon ring is recovered to at least 3 Å resolution. |
In addition to motion correction, the total dose used for one exposure can now be extended to greater than 60 e/Å 2 . Because the dose is divided by many frames, rather than one single image, the dose can be easily selected by summing a part of the frames. Electron radiation rapidly damages the high-frequency signal with the increasing of frame number, but less to the low-frequency signal. Therefore, the advantage of using a high dose is to increase image contrast which is contributed by low-frequency signal. High contrast will help to improve particle picking and 2D/3D alignment. In the final 3D reconstruction, the frames that are severely damaged by the radiation can be removed or low-pass filtered to remove the damaged high-frequency signals. [ 16 , 30 , 31 ] Such processing has been adopted in the final step of near-atomic resolution 3D reconstruction, and, typically, can gain the resolution with 0.1–0.5 Å.
Sample heterogeneity is always the first thing to be dealt with before obtaining a high-resolution 3D reconstruction by single-particle CryoEM. Besides the biochemistry purification, computational technology provides another way to isolate bio-macromolecules with different conformation or composition, termed classification. The classification can be performed at 2D and 3D levels. For 2D classification, it classifies the 2D images based on their similarities into many classes presumably different in projection direction, conformation or composition. The 2D classification is efficient for particle screening and isolating bad particles, and has been well established. For 3D classification, it is much more challenging than the 2D case due to the complexity of 3D alignment when heterogeneity is present. Owing to the low contrast of CryoEM images, it is very uncertain to recognize a different feature and assign the corresponding image to a certain class. Recently, a maximum-likelihood (ML) based method was developed and implemented with a user-friendly interface. [ 32 ] Different from the traditional method which assigns each particle image to a specific class, ML method calculates the probability of a particle image belonging to a class. Based on the probability, each particle is assigned and contributes to all classes but with different weighting. The ML method has been proved efficient by a series of high-resolution 3D reconstructions of biological macromolecules with small size and heterogeneity. [ 20 , 21 , 33 , 34 ]
Advanced by a series of recent breakthroughs in new technologies, including DDD and counting camera, movie image processing and ML-based classification, CryoEM is undergoing a resolution revolution [ 35 ] and is no longer a “blobiology” technology. [ 36 ] The technical advancements make CryoEM become a complementary tool to x-ray crystallography, especially for large protein complexes. [ 37 ] A series of important complexes and assemblies hard to crystallize have been solved in near atomic resolution by single-particle CryoEM. Throughout the history of CryoEM, technical advancements have always been the chief moving force, from cryosample preparation, parallel electron beam to the recent camera technology. The image processing technology and userfriendly software tools, including SPIDER, [ 38 ] EAMN [ 39 ] and Relion, [ 32 ] FREALIGN, [ 40 ] etc, also promoted broad applications of CryoEM in structural biology. However, although the feasibility of obtaining comparable resolution as x-ray crystallography has been proven by several structures determined at 2–3 Å resolution, [ 31 , 41 ] it is still a challenge to routinely realize such atomic-resolution structure determination for all suitable samples by CryoEM.
There are a few factors limiting the routine atomic resolution. First, biochemistry still plays the most important role in determining the atomic-resolution structure of a biomacromolecule by CryoEM. Homogeneity in conformation and composition is the chief factor that decides the final resolution, although the 3D classification approach is becoming more and more powerful and can partially deal with some local heterogeneity problems. Second, the computing efficiency of current CryoEM is still low. The classification algorithm usually requires hundreds of thousands or even millions of particle images to analyze in order to sort out the best images. Although the new cameras and corresponding dose fractionation technology significantly improve image quality, they output much larger image files, ∼2 gigabytes for an image with 30 frames, than traditional cameras. Combining the requirement for a huge number of images with large file size, the size of a CryoEM dataset is typically several terabytes and corresponding image processing usually takes several weeks or even months. Therefore, developing a more efficient computing algorithm and investing more clusters with massive high-performance storage and high-speed network are becoming more and more necessary for current and future CryoEM. Third, the entire procedure from data collection, image pre-processing to final 3D reconstruction and model building varies in a case-by-case manner and strongly depends on user experiences. A robust and fully automatic system combining all steps is still missing, which limits the wide application of atomic-resolution CryoEM.
In addition to the recent progresses described above, there are also many other new technologies being developed and applied to CryoEM. To improve computing efficiency, the general purpose graphic processing unit (GPU) has been introduced to accelerate CryoEM image processing and showed a speedup of ∼ 10–100 folds. [ 42 , 43 ] GPU is expected to be a good solution to improve computing efficiency with low cost and high density. Phase plate is a hardware designed to enhance the low-frequency contrast of the image. Small molecules less than 100–200 kDa are still a difficult task for CryoEM due to the low contrast. The phase plate potentially extends the capability of CryoEM to smaller complexes and assemblies. [ 44 – 47 ] Sample preparation techniques, such as graphene and gold supporting film, [ 48 , 49 ] are making the biological sample more stable under electron beam and easier to prepare for atomic resolution CryoEM. These technologies are driving CryoEM into a bright future and making it a routinelyused tool in more and more biology laboratories.
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 | |
| 22 | |
| 23 | |
| 24 | |
| 25 | |
| 26 | |
| 27 | |
| 28 | |
| 29 | |
| 30 | |
| 31 | |
| 32 | |
| 33 | |
| 34 | |
| 35 | |
| 36 | |
| 37 | |
| 38 | |
| 39 | |
| 40 | |
| 41 | |
| 42 | |
| 43 | |
| 44 | |
| 45 | |
| 46 | |
| 47 | |
| 48 | |
| 49 |