{kind=link}
{kind=link}
{kind=link}
Computational investigations on polymerase actions in gene transcription and replication: Combining physical modeling and atomistic simulations
[Yu Jin †,  ]
]
]
|
|


† Corresponding author. E-mail:
Project supported by the National Natural Science Foundation (Grant No. 11275022).
Polymerases are protein enzymes that move along nucleic acid chains and catalyze template-based polymerization reactions during gene transcription and replication. The polymerases also substantially improve transcription or replication fidelity through the non-equilibrium enzymatic cycles. We briefly review computational efforts that have been made toward understanding mechano–chemical coupling and fidelity control mechanisms of the polymerase elongation. The polymerases are regarded as molecular information motors during the elongation process. It requires a full spectrum of computational approaches from multiple time and length scales to understand the full polymerase functional cycle. We stay away from quantum mechanics based approaches to the polymerase catalysis due to abundant former surveys, while addressing statistical physics modeling approaches along with all-atom molecular dynamics simulation studies. We organize this review around our own modeling and simulation practices on a single subunit T7 RNA polymerase, and summarize commensurate studies on structurally similar DNA polymerases as well. For multi-subunit RNA polymerases that have been actively studied in recent years, we leave systematical reviews of the simulation achievements to latest computational chemistry surveys, while covering only representative studies published very recently, including our own work modeling structure-based elongation kinetic of yeast RNA polymerase II. In the end, we briefly go through physical modeling on elongation pauses and backtracking activities of the multi-subunit RNAPs. We emphasize on the fluctuation and control mechanisms of the polymerase actions, highlight the non-equilibrium nature of the operation system, and try to build some perspectives toward understanding the polymerase impacts from the single molecule level to a genome-wide scale.
Polymerases are key protein enzymes that direct gene transcription and replication in the central dogma of molecular biology. They move along the nucleic acid (NA) track as molecular motors [ 1 ] and catalyze RNA or DNA synthesis according to the template NA strand. The chemical catalysis, mechanical performance, and fidelity control of polymerases are therefore critical for maintaining human health, while the malfunctions lead to diverse genetic diseases. [ 2 , 3 ] The polymerase enzymes are widely utilized in synthetic gene expression systems [ 4 – 6 ] and in genomic technologies. [ 7 – 9 ] Engineering and redesigning of these enzymes are of high interest to various implementations. With technological advancements in tracking and manipulating polymerase enzymes at the single molecule level in recent years, [ 10 – 13 ] fundamental mechanisms underlying individual polymerase actions become approachable, which greatly improve our understandings and promote further applications.
To understand the functional mechanisms of polymerase enzymes with structural, dynamical, and energetic details, computational studies are indispensible. With rapid developments on high-performance computing using parallel computer clusters, [ 14 – 16 ] molecular dynamics (MD) simulations of protein macromolecules demonstrate great potential in elucidating the underlying mechanisms from the bottom up at atomistic resolution. [ 17 ] Along with improvements on the allatom force field, [ 18 ] long simulations approaching the microseconds to milliseconds physiological time scale have been achieved. [ 15 , 16 , 19 ] With further improvements on sampling techniques and data analysis methods, [ 20 – 24 ] simulations become much more efficient in evaluating energetics and other physiologically relevant quantities. On the other hand, a top down modeling strategy toward solving specific problems at commensurable scales, as commonly practiced in physical and mathematical sciences, can effectively deal with interested properties and easily connect to experimental measurements. In this article, we aim at providing a brief review, based on our own efforts and practices, combining both the top down and the bottom up computational strategies on studying the polymerase functions.
Without being able to survey a full spectrum of computational approaches on this topic, we focus only on stochastic or kinetic modeling studies from statistical physics perspective as well as from atomistic MD simulations that reveal mechano-chemical coupling and fidelity control properties of the polymerases. We start with a retrospect to early and general modeling frameworks built for the polymerase action, then emphasize on studies of single subunit DNA polymerases (DNAPs) and RNA polymerases (RNAPs) that are relatively simple in structures. For structurally more complex multisubunit RNAPs that are studied intensively in recent years, we refer readers to two wonderful reviews, [ 25 , 26 ] which present systematically the simulation studies. We then show a few of the latest representative works that possibly shed light on future studies of polymerase functional controls.
An early review on the single nucleotide addition cycle (NAC) of transcription provides a nice thermodynamics framework on RNA synthesis. [ 27 ] For each NAC, an incoming NTP is added to the existing RNA strand and the product pyrophosphate ion (PPi) is released: RNA i +NTP ⇔ RNA i +1 +PPi, the RNAP also moves from position i to i + 1. The corresponding Gibbs free energy change Δ G can be decomposed into three parts: a chemical part, an RNA transcript folding part, and an RNAP elongation complex part (including the double and single-stranded DNA in the transcription bubble). In particular, the chemical part is 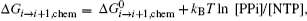
Inspired by single molecule measurements on the load force–velocity relationships of E. coli RNAP, an early mechanical model [ 29 ] treated the RNAP as a processive molecular motor capable of generating a force of 25–30 pN. The model assumed a rate-limiting step on the PPi release, and suggested that NTP binding rectifies RNAP diffusion on the DNA track. Although the assumption was not confirmed by later studies, the Brownian ratchet nature of the RNAP was captured nicely in that model. [ 29 ] Based on similar single molecule measurements, [ 30 ] another stochastic model of RNAP was built [ 31 ] with a focus on explaining the stall force distribution detected in the experiments. The model predicted that the stall force experimentally detected would be significantly smaller than the thermodynamic stall force. [ 31 ] In both models, DNA sequence-dependent effects had been introduced. These early modeling studies shed light on using chemical kinetics or stochastic methods to effectively describe the mechanochemical coupling in RNAPs.
There are a few more recent modeling approaches toward understanding the general RNAP properties. For example, a ‘look-ahead’ model for the transcription elongation has been proposed, in which NTP binds reversibly to a DNA site a few bps (∼ 4 bp) ahead before being incorporated covalently into the nascent RNA chain. [ 32 ] The model does not concern the mechanistic nature but provides a chemical kinetic framework, in which transcription fidelity control through NTP selection is performed at several DNA template sites simultaneously. [ 32 ] In another example, a general kinetic model was developed for the whole transcription cycle, taking into account that after RNA synthesis, RNAP may diffuse along DNA, desorb, or return to the promoter site to restart transcription. [ 33 ] Interestingly, the model can predict transcriptional bursts even in the absence of explicit regulation of the transcription by master proteins. [ 33 ] In a third example, the dwell-time distributions in a two-state motor model was derived first, and on top of that, an RNAP traffic model was developed considering steric interactions among many RNAPs moving simultaneously on the same track. [ 34 ] One more example we want to mention here is the development of a ‘modular’ scheme of the RNAP transcription kinetics, [ 35 ] which considers alternative and offpathway states (e.g. paused, backtracked, arrested, and terminated states) of the RNAP elongation complex. The framework can be extended to study DNA replication, repair, RNA translation, etc. [ 35 ]
Below we focus on a group of single subunit polymerases [ 36 – 38 ] which include both DNA and RNA polymerases for gene replication and transcription, respectively. These polymerases adopt similar hand-like structures and are connected evolutionarily. We first go through studies examining mechano–chemical coupling properties of the system. These studies mainly rely on molecular modeling and simulation techniques. Then we address how fidelity control is achieved at the substrate selection stage, which has been studied from both molecular simulation and non-equilibrium statistic physics perspectives.
Since polymerases work as molecular motors, we concern about how chemical free energy is transformed into mechanical work during each NAC cycle. The chemical free energy (Δ G i → i +1,checm ) basically supports the phosphoryl transfer reaction that adds the NMP part of NTP to the existing RNA strand while it dissociates the PPi part. At the same time during each NAC, the polymerase undergoes substantial conformational changes to allow NTP binding and insertion; then it recovers back to the initial conformation, during or after the PPi release and translocation. Correspondingly, the mechanical motions involve both the substantial conformational changes of the polymerase and the relative translocation between the polymerase and the NA track. As a molecular motor moving along the track, the most concerned mechano– chemical coupling feature is whether the polymerase translocation is directly coupled to the chemical step during the enzymatic cycle from the NTP binding to the PPi dissociation. Besides, the translocation may also couple to part of the substantial conformational changes. Indeed, a previous highresolution structural study on bacteriophage T7 RNAP suggested a power stroke mechanism, [ 39 ] in which the PPi release is tightly coupled to the translocation through an O-helix or fingers domain opening motion. Under this mechanism, the PPi release energetically supports or drives the translocation.
An all-atom MD simulation study was conducted on T7 RNAP, examining the energetics of the translocation. [ 40 ] The MD study indicated that without the fingers domain opening after the product release, the translocation is not preferred. Though large fluctuations of the RNA 3’-end were detected within the nanosecond simulation, large conformational changes and critical translocation could not be sampled. The translocation mechanism of a structurally similar DNA polymerase I (pol I) from Bacillus stearothermophilus was also studied by all-atom MD simulations, employing biased and targeted MD methods. [ 41 ] The study demonstrated that the PPi release precedes the translocation and facilitates the fingers domain opening transition, which is then followed by DNA displacements for the translocation. Both studies suggested that the translocation of the polymerase is coupled to the domain opening conformational transition.
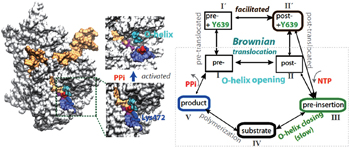
In our most recent MD studies on the PPi release of T7 RNAP, [ 42 ] we constructed the Markov state model (MSM) using a large number of nanosecond simulations, as that performed for the multi-subunit RNAPs. [ 43 – 45 ] In addition, we also conducted a small number of microsecond simulations to detect slow motions in the PPi release process. In contrast with the charge-facilitated model of the PPi release discovered in the multi-subunit RNAPs, [ 43 – 45 ] it was found that the PPi release in the single-subunit T7 RNAP proceeds through a ‘jump-from-cavity’ activation process, assisted by a large swing of side chain Lys472 (see Fig.
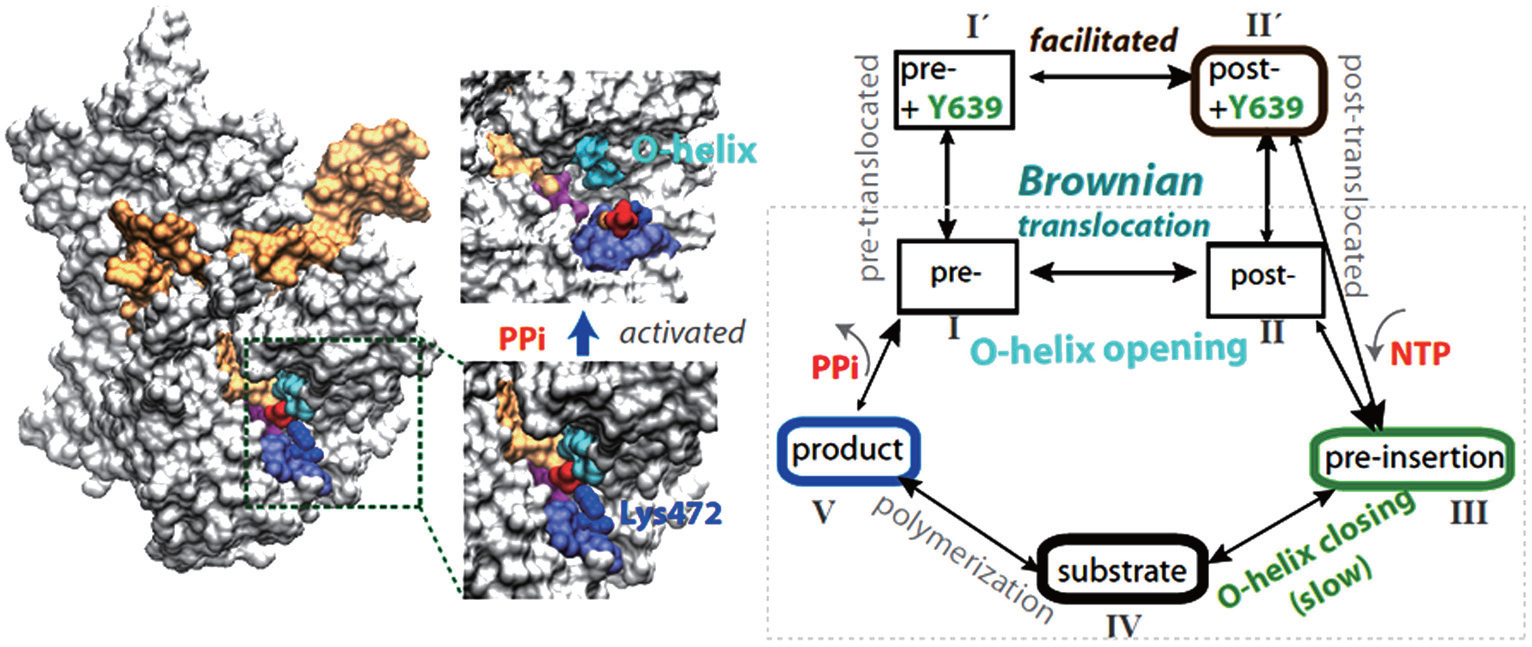 | Fig. 1. Transcription elongation of T7 RNAP and mechano–chemical coupling features. (a) The product structure of single subunit T7 RNAP elongation complex (in a surface representation: protein, white; NA, orange; the O-helix on the fingers domain, cyan; PPi, red; Lys472, blue, and the linked loop, light blue). The PPi release is found to be a jump-from-cavity process that is assisted by Lys472 side chain swing. [ 42 ] The release does not appear to be tightly coupled to the O-helix or the fingers domain opening, thus, cannot drive the translocation. (b) A kinetic scheme of T7 elongation used in our recent modeling work, [ 53 ] from NTP binding (pre-insertion) and insertion (the fingers domain or O-helix closing) to chemical reaction and product (PPi) release, followed by translocation. The translocation proceeds in Brownian movements, while the NTP binding serves for a pawl in the Brownian ratchet scenario to prevent backward movements. A small post-translocation free energy bias has been suggested to stabilize Y639 for the incoming nucleotide selection. [ 53 ] |
From the above analyses, one can see that the substantial conformational changes in regard to the fingers domain opening and closing are crucial for the functioning of the single subunit polymerases. Recently, the domain opening process of DNA pol I has been directly simulated using microsecond unbiased MDs at atomistic resolution. [ 54 ] An ‘ajar’ (semi-open) intermediate conformation, which had been discovered from a mismatched nucleotide bound structure, [ 55 ] was examined in the simulation as well, and four backbone dihedrals were identified as being important for the opening process.
While the opening conformational transition after the PPi release somehow couples to the translocation, the transition after the NTP binding accompanies the NTP insertion to the active site, which can be a rate limiting process in some polymerases. The domain opening/closing motions have been examined previously through elastic network models combining with normal mode analyses. [ 56 , 57 ] It was noticed that the open to the closed state transition could be well approximated by a small number of normal modes of the open form polymerase. [ 56 ] Later, a network of residues spanning the flexible fingers domain and the stable palm domain were found to be involved in the open-closed transition, and the conserved network of residues supported a common induced-fit mechanism in the polymerase families for the closed structure formation. [ 57 ] A comprehensive report was made recently toward understanding the pre-chemistry open-closed conformational changes in eukaryotic DNA polymerase β . [ 58 ] The NTP substrate induced the domain closing transition in particular assembles the polymerase active site prior to chemistry, contributing essentially to DNA synthesis as well as to fidelity. [ 58 ] The potential of mean force for the pol β closing pathway prior to chemistry was demonstrated in the study without NTP, as well as in the presence of correct and incorrect NTPs. [ 58 ] It was shown that the subdomain motions appear to be intrinsic (as for conformational selection), subtle side chain motions and their favored states are largely determined by the binding of the substrate (as for induced fit). Hence, a hybrid of the conformational selection and the induced fit mechanisms seems to apply to DNA polymerases. [ 58 ]
It is generally assumed that the polymerase fidelity control is achieved through both NTP binding and chemical steps. Some of the single subunit DNAPs, such as T7 DNAP and eukaryotic DNA pol β , have been studied systematically. For example, relative stabilities of Watson–Crick and mismatched dNTP* template base pairs in the active site of T7 DNAP and human DNA pol β have been examined using MD simulations and linear-response analyses. [ 59 , 60 ] It was found that the NTP binding selectivity of T7 DNAP is largely determined by the template–NTP interaction, while the binding contribution toward the replication fidelity control is less significant in pol β than that in T7 DNAP. Further progress toward understanding the fidelity control of these two types of DNAPs can be found, for example, in Refs. [ 61 ]–[ 63 ]. In Ref. [ 61 ], a variety of computational methods, including the free energy perturbation, the linear response approximation, and the empirical valence bond method were summarized for calculating the binding free energy contribution. In Ref. [ 62 ], the full fidelity control of T7 DNAP was studied for both the substrate binding and the chemical step by taking into account contributions from the binding, pKa shifts, PO bonding breaking and making. More recently, a binding free energy decomposition approach aiming at an accurate quantification of the pol β fidelity control was implemented, [ 63 ] in which separate calculations on the neutral base and charged phosphate part using different dielectric constants were conducted.
As a small eukaryotic enzyme being able to repair short single stranded DNA, pol β has been extensively studied on its fidelity control. Beside the binding free energy, the closed to open transition of pol β was examined by targeted MDsimulations in the mismatched system in order to explain experimental results regarding inefficient DNA extension following misincorporation, or polymerase proofreading. [ 64 ] Recently, the crystal structures of pol β bound with the mismatched NTPs have been reported, [ 65 ] together with MD simulation elucidating the replication fidelity control at both the open and closed conformations. The results clearly showed different DNAP responses toward different mismatches. Simulation studies on fidelity control of other single subunit polymerases also emerged recently. For example, MD studies on viral RNAdependent RNA polymerases (RdRps) revealed coevolution dynamics with structure and function suggested by the conserved and correlated dynamics of fidelity control and structural elements. [ 66 ] More recently, the RdRp from Poliovirus was studied through MD simulations and using free energy calculation. [ 67 ] Interestingly, the dynamic correlation between two important motifs appears sensitive to the incoming NTP species; the accessibility of the active site by one of the motifs also depends on the base pairing strength between the incoming NTP and the template; it explains why the active-site closure can be triggered by a correct NTP. [ 67 ] Furthermore, studies on HIV reverse transcriptase have been conducted both experimentally and computationally. [ 68 ] The studies showed that the initial steps of weak substrate binding and protein conformational transition significantly enrich the yield of a reaction of a correct substrate but diminish that for an incorrect one.
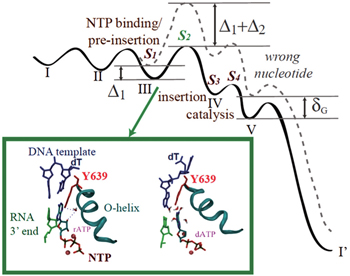
Among those studies above, controversies arose, for example, on how much pre-chemistry and chemical steps contribute to the DNAP replication fidelity control. [ 58 , 69 , 70 ] Recently, we put up a kinetic framework on analyzing the stepwise nucleotide selection in the polymerase elongation, [ 71 ] which considers contributions to the fidelity control from each kinetic checkpoint before the end of the catalysis. When the elongation kinetics is described well by a three-state model (consisting of NTP binding, catalysis, and translocation), the nucleotide selection can happen at two checkpoints, i.e., upon NTP binding and during the chemistry step, as pointed out early. When the polymerase elongation cycle is detected with more intermediate states, however, additional kinetic transitions and checkpoints should be included. For example, NTP binding/pre-insertion can be followed by another pre-chemistry step, which allows the NTP insertion along with an open to closed conformational transition (see Fig.
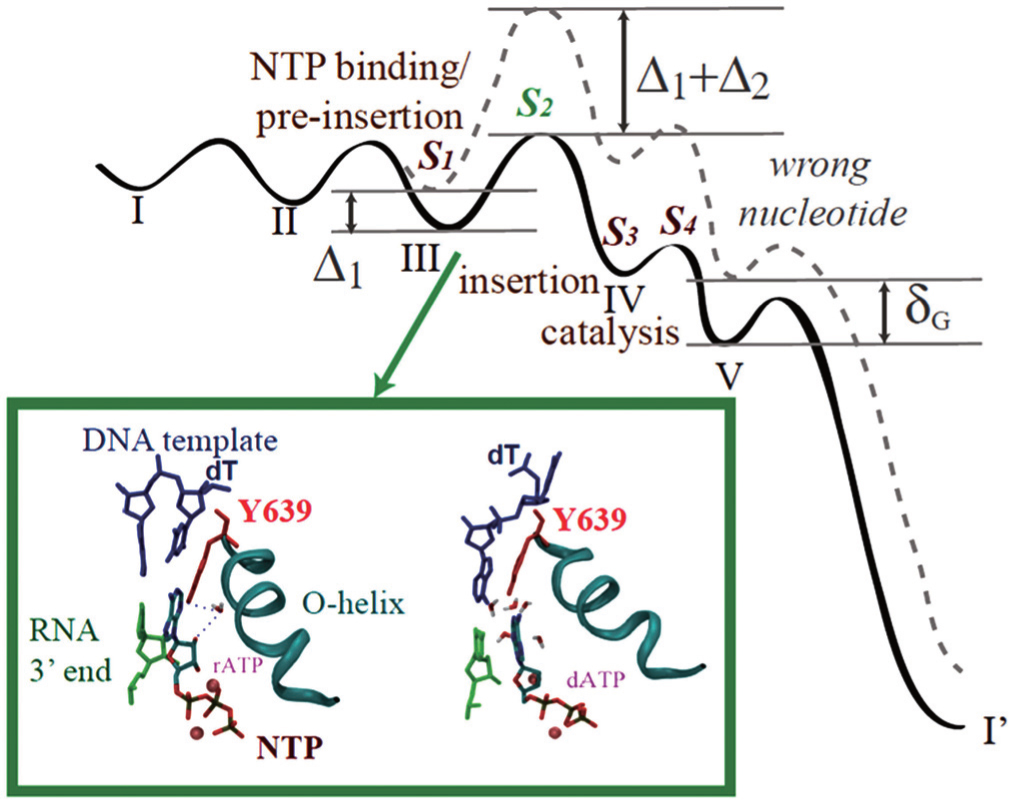 | Fig. 2. The nucleotide selection scheme in T7 RNAP elongation. (Top) The free energy landscape for incorporating the right (solid line) and wrong (dashed) nucleotides in the five-state kinetic scheme. Four selection checkpoints ( S 1 to S 4 ) are labeled. Δ 1 and Δ 2 are differentiation free energies between the right and wrong nucleotides at the first two checkpoints. δ G is an overall free energy differentiation without polymerase. See Ref. [ 71 ] for details. (Bottom) Comparing the active site configurations when the right and wrong NTP bind respectively to the pre-insertion site. [ 72 ] Y639 (red) is located on the C-term end of the Ohelix (cyan) to assist the nucleotide selection. Left: rATP (right) forms the Watson–Crick base pairing with the template. The recognition is assisted by water bridging HB interactions with Y639-OH and 2’-OH of rNTP. Right: dATP cannot base pair with the template due to the Y639 interference, which associates with dATP and stacks well with the DNA–RNA hybrid end, under water collision. [ 72 ] |
Using the master equation approach, we demonstrated some interesting properties in the stepwise selection system. First, we notice that the selection through the initial checkpoint ( S 1 ), i.e., rejecting the wrong nucleotides immediately upon binding/pre-insertion, keeps the elongation at a relative high speed, which would not be maintained if the initial screening is not conducted.
Next, we find that for the same amount of free energy differentiation Δ, the same error rate is achieved between the neighboring rejection and inhibition (i.e., S 1 and S 2 , or S 3 and S 4 ). Finally, we show that the error rate achieved under the early checkpoints ( S 1 and S 2 ) is usually lower than that achieved later on the reaction pathway ( S 3 and S 4 ), if the same amount of free energy differentiation applies. One can then systematically characterize the stepwise selection, i.e., by calculating the differentiation free energy at each checkpoint. We are now performing the analyses on T7 RNAP to see if the selection system is evolved to be sufficiently efficient in the absence of proofreading.
Besides, we have performedMDsimulations to T7 RNAP and found a critical residue Tyr639 that assists nucleotide selection from pre-insertion to insertion. [ 72 ] This residue is marginally stabilized inside the active site at post-translocation by stacking its side chain with the end bp of the DNA–RNA hybrid. A cognate rNTP (rATP, see Fig.
Furthermore, one should bear in mind that the polymerase elongation is a non-equilibrium process, and there are theoretical and modeling efforts made along this direction. It is understood that the equilibrium free energy difference between the right and wrong nucleotide incorporations contributes to transcription or replication fidelity, but the contribution is too small to account for the overall fidelity. The template-based non-equilibrium copolymerization process has been analyzed focusing on interplay between information acquisition and thermodynamic driving force. [ 74 , 75 ] It is clearly shown that the polymerase must operate far from equilibrium to achieve a high fidelity level. Interestingly, close to equilibrium, the polymerase growth or elongation can be essentially supported by configuration disorder or the incorporation of ‘errors’. [ 74 ] The open-system thermodynamics to achieve DNA polymerase fidelity was systematically analyzed in Ref. [ 76 ]. In particular, the nucleotide insertion selection in the absence of the exonuclease proofreading has been considered. The study indicated that a sustained non-equilibrium steady state essentially drives the polymerization error rate to transit from a thermodynamically determined value to a kinetically determined one, i.e., relatively high fidelity is achieved under the “flux-driven kinetic checkpoints”. [ 76 ] The two discrimination mechanisms involving either energetic (different binding energies) or kinetic (different kinetic barriers) differentiation were also analyzed more recently in Ref. .[76]. It was shown that the two mechanisms cannot be mixed in a single-step reaction to reduce errors, but they can be combined in coping schemes with error correction through proofreading. [ 77 ] As a matter of fact, we have not been aware of systematical computational investigations on the DNAP proofreading activities, which involve the polymerase active site switching to the exonuclease cleavage site to allow excision on the mis-incorporation. [ 78 ]
Multi-subunit RNA polymerases (RNAPs) are widely distributed from bacteria to higher organisms, and have been extensively studied. [ 79 – 82 ] Besides those general models, early kinetic modeling and analyses on multi-subunit RNAPs were developed side by side with single molecule experiments. [ 49 , 83 , 84 ] For example, through a combination of theoretical and experimental approaches, a sequencedependent thermal ratchet model of the transcription elongation was built. [ 85 ] The NTP-specific model parameters were also obtained according to the force–velocity measurements on E. coli RNAP. [ 85 ] A continuum Fokker–Planck framework of the RNAP elongation was developed as well. [ 86 ] In particular, using high-resolution single-molecule data [ 49 ] of E. coli RNAP at the near equilibrium condition, a free energy profile of the polymerase translocation was obtained, [ 86 ] which shows consistently the ratchet character of the RNAP elongation. Stationary distributions of the RNAP translocation at farfrom- equilibrium condition (e.g. very high [NTP]) can also be derived under this framework. [ 86 ] For multi-subunit RNAPs, we also cover two types of computational studies: one is on structure-based modeling and simulations, concerning molecular details of internal coupling and control; the other is on statistical or kinetic modeling, with a focus on backtracking, pauses, and related proofreading activities.
Though both high-resolution structural studies and single molecule force measurements have been extensively conducted on multi-subunit RNAPs, there is still lack of detailed structural dynamics. Nevertheless, the dynamical detail can be probed directly from the ‘computational microscope’ at atomistic resolution. As mentioned earlier, systematical reviews on employing MD simulation methods to study the multi-subunit RNAPs can be found in Refs. [ 25 ] and [ 26 ]. Here we only introduce representative works published very recently, which have not been included in the above reviews.
In regard to the mechano–chemical coupling of RNAP, a central concern is the translocation mechanism. The translocation is studied intensively by constructing the Markov state model (MSM) for yeast Pol II based on a large number of short (nanoseconds) MD simulations. [ 87 ] The simulation system of Pol II reaches close to a half million atoms in the explicit solvent condition. It is a big challenge, indeed, to simulate a molecular machine like Pol II to biologically relevant time scales from micro to milliseconds. Launching many short simulations scattered around the conformational space improves the sampling efficiency, while constructing the MSM essentially extracts the kinetic information from the simulated data in the high-dimensional space. The studies show that the Pol II translocation is driven thermally. [ 87 ] In particular, metastable intermediate states between the pre- and post-translocation states have been identified. It is also found that fluctuations of a bridge helix between the bent and straight conformations facilitate the translocation of the upstream RNA:DNA hybrid, which turns out to be a rate-limiting step of the translocation. The bridge helix fluctuations also facilitate the translocation of a ‘transition nucleotide’, which moves asynchronously from the rest of the upstream RNA and DNA in the hybrid region. [ 87 ] According to the MSM, the overall translocation rate is estimated to be about tens of microseconds at least, which can be very fast compared to the rate-limiting step of the elongation cycle (tens of milliseconds). Anyhow, including the full transcription bubble may slow down the translocation rate. [ 88 ] In addition, it has been noted that the translocation is simulated with a trigger loop (TL) in an open conformation already. [ 87 ] The TL open has been suggested to be a pre-requisite for the translocation to happen. [ 89 , 90 ]
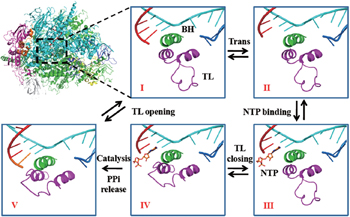
Interestingly, recent single molecule experiments on Pol II identified a slow force-dependent step in the Pol II elongation, aside from a rate-limiting force-independent transition. [ 91 ] Considering that the TL opening motion can be slow as well as force dependent, we built a structure-based kinetic model of Pol II elongation [ 92 ] (see Fig.
 | Fig. 3. A proposed five-state Brownian ratchet model of the multi-subunit RNA polymerase II (Pol II) elongation, adopted from Ref. [ 92 ]. The structure of Pol II is provided (upper left). Configurations of the trigger loop (TL, in purple) and bridge helix (BH, in green) around the active site are shown for five kinetic states (I to V) in five windows. The non-template DNA strand is shown in blue, the template DNA strand is shown in cyan, and the synthesizing RNA strand is shown in red. The incoming NTP molecule is shown in orange. |
In regard to the fidelity control, systematical ‘five checkpoints’ mechanisms were presented implementing MD simulations. [ 93 ] In the multi-subunit RNAP, there is an entry site ( E -site) for the NTP binding prior to the NTP insertion into the active site ( A -site). Correspondingly, the five checkpoints follow the reaction path along the elongation cycle, as the initial NTP binding to the E -site, a transition or rotation of NTP from the E -site to the A -site ( E – A rotation), the TL closing, active site re-arrangement for catalysis, and finally, the backtracking. [ 93 ] The first four checkpoints are indeed for the NTP selection, while the last checkpoint serves for proofreading. In particular, the umbrella sampling method was implemented to calculate the free energy against the mismatched NTP binding at the first checkpoint when the TL is still open. [ 93 ] The studies also found that the most important checkpoint for deoxy-NTP discrimination happens when the mismatched dNTP triggers conformational distortions in the active site to hinder the catalysis. [ 93 ] The final checkpoint to trigger the backtracking is through distortions of the template DNA nucleotide and DNA–RNA hybrid base pair around the active site. The studies open the door for very detailed studies on the fidelity control mechanisms. It is noted, as the authors pointed out, that the efficiencies of the fidelity checkpoints on discriminating against different non-cognate rNTPs and dNTPs are sequence dependent and vary for different RNAP species.
In multi-subunit RNAPs, pauses are frequently present to play important regulation roles. [ 94 ] The paused states are often linked to backtracking behaviors of the polymerases. [ 95 , 96 ] A statistical mechanics approach toward predicting backtracked pauses in bacterial transcription elongation has been successfully applied. [ 97 ] A thermodynamic model of the elongation complex was built with sequence-dependent free energy variations from the translocational and size fluctuations of the transcription bubble, as well as from accompanied changes in the RNA–RNA hybrid and the RNA transcript. The model produced statistically significant results toward predicting ∼ 100 elongation pause sites for E. coli RNAP on 10 DNA templates. [ 97 ] The study also provided a kinetic model on pause recovery, assuming slow RNA unfolding and fast translocation. In these models, the sequence-specific kinetic barriers due to RNA co-transcriptional folding turn out to be essential to strongly inhibit the backtracking. Another essential feature identified is an intermediate state separating the productive elongation with the backtracking in a further developed thermal ratchet model. [ 98 ] Whether the backtracking causes a wide range of pauses, including both long and short ones, was also investigated. [ 95 ] By modeling the backtracking as force-biased diffusion on a periodic one-dimension free energy landscape, the study showed that a single mechanism of random walk backtracking can generate both the long (diffusive) and short (ubiquitous) pauses. In particular for the short pauses, sequence-induced variations on the backward rates can have a large impact on the lifetime of the backtracking pauses. [ 95 ] Actually, when the backtracking pauses are included in a full transcription elongation model, a broad, heavytailed distribution of the elongation time has been obtained. [ 99 ] Interestingly, the authors of the study suggested that the pauses could even lead to bursts of mRNA production and non- Poisson statistics of mRNA levels, [ 99 ] thus, contribute significantly to noise productions on a cellular level. Using a similar kinetic model and the master equation approach, these researchers studied proofreading activities involving the backtracking and RNA cleavage. [ 100 ] Backtracking by more than one nucleotide provides a multiple-checking reaction to probe the fidelity of newly generated nucleotides before further nucleotide addition. The study showed that the accuracy improves along with longer delay caused by the backtracking and cleavage. In an extreme case, the error fraction scales exponentially with the maximum backtracking distances. [ 100 ] The model thus predicted a strong dependence of transcriptional fidelity on the backtracking rates or probabilities. More recently, some exact results for a kinetic model of backtracking were presented, along with analyses of the impact on the speed and accuracy of the transcription. [ 101 ] On top of these kinetic models, it is expected that more detailed models on backtracking can be developed, connecting specific responses and control mechanisms with identified structure features of the RNAPs.
Gene transcription and replication are directed by RNA and DNA polymerases through their enzymatic cycles. Correspondingly, the elongation processes are maintained at the non-equilibrium steady state (NESS) driven by the chemical potential. [ 102 ] It is key to understand the NESS basis in order to understand the mechano–chemical coupling mechanisms and fidelity control features of the polymerases. The non-equilibrium statistical physics has been built up recently in Ref. [ 103 ], which sets the minimum value of the physically allowed rate of heat production in terms of the growth rate, internal entropy, and durability of the ‘self-replication’: the more irreversible the macroscopic process, the more positive the minimum entropy production. Nevertheless, closeto- equilibrium properties of individual kinetic states in the elongation cycle can still be probed well through equilibrium MD simulations, so that local relaxation dynamics and energetics reveal substantial structural details. For rate-limiting conformational transitions in the elongation cycle, however, commensurable simulations are expected to take into account the NESS chemical potential by simulating sufficiently fast processes of substrate binding and product release. The non-equilibrium setup has not been implemented in regular nanosecond MD simulation studies of the enzyme and molecular machines. The NESS dynamics would then become more of a concern when micro to milliseconds MD simulations become routine in the future.
It is quite interesting to note that polymerases have been largely identified to work under the loosely coupled Brownian ratchet scenario, no matter whether for the single or multi-subunit polymerases. In addition to abundant experimental evidences revealed early, a recently detected example is on the translocation of replicative DNAP from bacteriophage phi29. [ 104 ] Under the Brownian ratchet mechanism, the translocation of the polymerase spontaneously happens without being directly coupled to a chemical transition such as the substrate binding or product release. However, the translocation can still couple with some essential conformational changes (such as the O-helix or TL opening in the single and multi-subunit polymerases, respectively), which may be facilitated by the chemical transition but not at the same time during the coupling period with the translocation. In contrast, the tightly coupled power stroke scenario requires direct coupling between the translocation and the chemical transition, no matter other conformational changes involved or not. The power stroke scenario, however, has not gained continuous experimental support. Besides for the polymerases, ribosomes, the essential translation machinery, have also been consistently demonstrated to work under the Brownian ratchet scenario. [ 105 – 107 ] Since both machineries appeared very early in the molecular evolution history, one would speculate that the Brownian ratchet requires no highly sophisticated internal coupling mechanisms, therefore, might be easily adopted into those ancient molecular machineries. Systematical investigations on evolutionarily connected mechano–chemical coupling properties of the molecular machines are yet to be conducted.
The fidelity control of polymerase transcription and replication is achieved in general by combining nucleotide selection before the catalysis with proofreading cleavage after the catalysis. Both mechanisms work at non-equilibrium or driven conditions. The selection proceeds stepwise through each kinetic intermediate state, starting right after the nucleotide binding or pre-insertion, and working all the way until the end of the catalytic reaction. Substantial selection has been found to happen through the slow process of nucleotide insertion or catalysis. [ 58 , 70 ] We notice that early selections outperform the late ones on the reaction path in reducing the error rate while the initial selection or screening is particularly helpful to maintain the elongation speed high. Since the template-based polymerization relies primarily on the WC base pairing, the differentiation between incoming rNTP and dNTP becomes highly subtle, involving delicate residue coordination such as ‘steric gate’ or hydroxyl-water interaction etc. [ 72 , 108 ] On the other hand, tolerance on template backbone sugar heterogeneity is revealed, for example, for Pol II. [ 109 ] There are also evidences that the WC hydrogen bonding is not highly crucial for the fidelity control of T7 RNAP while the steric effect can be significant. [ 110 ] Remarkably, it was recently reported that transient WC-like mispairs (with probabilities 10 −3 –10 −5 ) stereochemically mimic the WC geometry so that they evade fidelity checkpoints, [ 111 ] which can play some universal role in gene mutation and molecular evolution. One may hypothesize that the transient WC-like mispairs set a limit on the polymerase fidelity control. Anyhow, it remains elusive how much the polymerases contribute energetically to select cognates over noncognates, especially, for variant nucleotide species. Hence, it is still hard to quantitatively test the hypothesis.
The original idea of kinetic proofreading traces back to work of Hopfield [ 112 ] and Ninio. [ 113 ] The proofreading activities of RNA polymerases have been investigated in recent years. [ 82 ] Interestingly, in a recently published modeling work, it was found that the proofreading supported fidelity control strongly depends on the sequence context such that it brings to accuracy variation to several orders of magnitude on a genome wide scale. [ 114 ] Though experimentally measured free energies of dsDNA and RNA–DNA hybrid have been incorporated into the model, the polymerase contribution to the sequencedependent accuracy variation has not been considered. [ 114 ] Again, it is because the polymerase contribution to the accuracy has not been systematically investigated. Hence, it becomes highly desirable if computational studies in the near future could quantify how much the polymerases energetically differentiate cognates vs. non-cognates, in a sequence specific manner. The sequence specific characterization of polymerase fidelity control and other actions can be gradually elucidated with technology advancements on targeted and genome wide sequencing. [ 7 , 115 ] After all, the complete functional cycles of the polymerases, including initiation, elongation, and termination, constantly involve mechanical responses of the protein machines toward sequence signals encoded in the genome as well as on further epigenetic modifications.
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 | |
| 22 | |
| 23 | |
| 24 | |
| 25 | |
| 26 | |
| 27 | |
| 28 | |
| 29 | |
| 30 | |
| 31 | |
| 32 | |
| 33 | |
| 34 | |
| 35 | |
| 36 | |
| 37 | |
| 38 | |
| 39 | |
| 40 | |
| 41 | |
| 42 | |
| 43 | |
| 44 | |
| 45 | |
| 46 | |
| 47 | |
| 48 | |
| 49 | |
| 50 | |
| 51 | |
| 52 | |
| 53 | |
| 54 | |
| 55 | |
| 56 | |
| 57 | |
| 58 | |
| 59 | |
| 60 | |
| 61 | |
| 62 | |
| 63 | |
| 64 | |
| 65 | |
| 66 | |
| 67 | |
| 68 | |
| 69 | |
| 70 | |
| 71 | |
| 72 | |
| 73 | |
| 74 | |
| 75 | |
| 76 | |
| 77 | |
| 78 | |
| 79 | |
| 80 | |
| 81 | |
| 82 | |
| 83 | |
| 84 | |
| 85 | |
| 86 | |
| 87 | |
| 88 | |
| 89 | |
| 90 | |
| 91 | |
| 92 | |
| 93 | |
| 94 | |
| 95 | |
| 96 | |
| 97 | |
| 98 | |
| 99 | |
| 100 | |
| 101 | |
| 102 | |
| 103 | |
| 104 | |
| 105 | |
| 106 | |
| 107 | |
| 108 | |
| 109 | |
| 110 | |
| 111 | |
| 112 | |
| 113 | |
| 114 | |
| 115 |