





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Surface structure evolution of cathode materials for Li-ion batteries
[Lyu Yingchun 1 , Liu Yali 2 , Gu Lin 1, 3, †,  ]
]
]
|
|


† Corresponding author. E-mail:
Project supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB07030200) and the National Basic Research Program of China (Grant Nos. 2014CB921002 and 2012CB921702).
Lithium ion batteries are important electrochemical energy storage devices for consumer electronics and the most promising candidates for electrical/hybrid vehicles. The surface chemistry influences the performance of the batteries significantly. In this short review, the evolution of the surface structure of the cathode materials at different states of the pristine, storage and electrochemical reactions are summarized. The main methods for the surface modification are also introduced.
Since lithium ion batteries were first commercialized in 1991 by Sony, they have been widely used in many fields such as consumer electronics, for their high energy density and high safety performance. [ 1 , 2 ] During the last 24 years, the performance of lithium ion batteries has significantly improved. The energy density has increased from 100 Wh·kg −1 to 260 Wh·kg −1 . Now, with emerging energy storage applications including electric vehicles (EVs), hybrid electric vehicles (HEVs) and grid, higher requirements are proposed for Li-ion batteries. [ 3 , 4 ]
Figure
 | Fig. 1. Typical Li-ion battery electrodes. (a) Low magnification (on left) and high magnification (inset on the right). (b) Schematics showing the main phases constituting a modern insertion cathode and their roles in transport. The polymeric binder is not shown. [ 5 ] |
A stable surface is a basic requirement for a perfect cathode. Film technologies offer good model electrodes to understand the influence of orientation, strain and stress on the electrochemical performance. In layered materials, the (001) plane exhibits higher discharge capacities than the (010) plane. An irreversible phase change occurs at the (010) surface, during the first (de)intercalation process, whereas reversible structural changes take place at the (001) surface. [ 6 ]
Actual surfaces usually contain defects, such as antisites, size effects, surface coating layers, gradient structure and core-shell structure. They may influence the surface structure, and introduce point defects and grain boundaries etc. to the structure. The types of defects and the concentration of the defects are controlled by thermodynamics. They are influenced by the components, crystal structure, and the synthesis methods. The defects may influence the transfer and storage of electrons and ions at the bulk, interface and surface. Defects can also offer extra sites for lithium storage. [ 7 – 9 ] The storage of lithium at the defects can be either reversible or irreversible. Among the cases, underpotential deposition mechanism, interfacial charging mechanism and surface charging mechanism are reversible. [ 10 ] The dangling bonds and impurity phase at the surface are irreversible. [ 10 ] Understanding the surface defect chemistry and control the surface defects enables regulating and controlling the transport, storage and reaction performance of the cathode.
Antisite defect is the most common defect in cathode materials. It has different influences on cathode materials based on the lithium diffusion channels. In materials with the one-dimensional (1D) channel, such as Li M PO 4 ( M = Fe, Co, Mn), the antisite defects strongly affect the electrochemical performance of the cathode material, since it will block the well-known one-dimensional lithium diffusion channel. [ 11 – 16 ] Controlling the antisite defects has been suggested as an effective way to improve the electrochemical performance of the olivine-type phosphates. [ 17 – 19 ] In materials with three-dimensional (3D) lithium diffusion channels, the antisite defects in spinel LiMn 2 O 4 increase the charge transfer impedance. [ 20 ] While antisite defects in triplite (Li 0.5 Fe 0.5 ) 2 SO 4 F does not block the Li + diffusion, it still exhibits an excellent electrochemical activity. [ 21 , 22 ]
In layered cathode materials, with 2D channel, antisite defect is the origin of the poor reactivity of LiNiO 2 and LiCrO 2 . [ 23 – 27 ] Our recent investigation of well-crystallized LiCrO 2 cathode indicates that Cr ions can migrate into the Li layers in the surface regions, leading to irreversible phase transformation from the layered to the rock-salt structure. The existence of the rock-salt phase in the surface blocks the extraction of lithium in the material. [ 27 ] LiNi 1/2 Mn 1/2 O 2 with fewer antisite defects can perform a much better property during cycle process. [ 25 , 28 ] Jinhyuk Lee et al. recently reported a Li-rich layered material which has a certain cation mixing but exhibits excellent performance. [ 29 , 30 ] In this layered material, Li diffuses from site to site by hopping through intermediate tetrahedral sites. Studies of Li 1.2 Cr 0.4 Mn 0.4 O 2 also show that cation mixing in lithium rich layered cathode does not affect the lithium storage capacity but stabilizes the structure. [ 31 ] Moreover, Li 3 NbO 4 -based system with cation-disordered rock-salt structure also shows a high capacity of approximately 300 mAh·g −1 , but the cycle performance is not acceptable. [ 32 ]
Size effects are also important. Nano-sized materials usually have a high rate performance, since the lithium ion diffusion path is much shorter in the nano materials. Micro-sized LiCrO 2 shows almost no signs of reversible lithium intercalation. [ 33 , 34 ] Feng et al. have found that the electrochemical cycling performance of LiCrO 2 is dependent on its particle size. If the grain size is reduced to nano-size, e.g. less than 20 nm, up to 0.7–0.9 lithium can be de-intercalated from the cathode and 0.5 lithium can be intercalated back into the cathode in the first cycle. [ 35 ] But nano-materials have a bigger specific surface area with more surface defect density, with higher surface energy. This usually causes serious side reaction during cycle. [ 36 , 37 ]
Other kinds of surface defects are lithium-deficiency, element enrichment, inhomogeneity and amorphousness. For example, Li 1.2 Ni 0.2 Mn 0.6 O 2 prepared by the hydrothermally assisted method have minimal Ni-rich surfaces and demonstrate much better capacity retention and much smaller voltage fade during cycling, compared to samples synthesized by coprecipitation or sol–gel methods. [ 38 , 39 ] The surface coating layers and gradient structures will be discussed in detail below.
The storage characteristics have very significant impacts on their structure and electrochemical performance. [ 40 – 46 ] Xia et al. [ 44 ] reported that commercial LiFePO 4 could be oxidized in humid and/or hot air and become α -Fe 2 O 3 and FePO 4 at the surface. A Li3PO 4 layer has been found at the LiFePO 4 grains’ surface after immersion in water. [ 40 ] Martin et al. [ 47 ] reported that the performance of LiFePO 4 /C nano-composites exposed to atmosphere at 120 °C decreases due to oxidation, involves H 2 O and the formation of disordered ferric lithium phosphate phase.
Li 2 CO 3 is commonly found on the surfaces of cathode materials that have been exposed to air. During storage, a thin layer forms on the particle surface and becomes thicker. Using LiNi 0.8 Co 0.15 Al 0.05 O 2 as an example, the formation of Li 2 CO 3 is presumed to take place via reaction


Figures
 | Fig. 2. TEM images of (a) fresh LiNi 0.8 Co 0.15 Al 0.05 O 2 powder and (b) LiNi 0.8 Co 0.15 Al 0.05 O 2 grain from air-exposed electrode. Charge and discharge profiles for (c) fresh cathode and (d) air-exposed cathode. [ 41 ] |
An ideal surface should be stable while soaked in the electrolyte and during the electrochemical process. But the actual surfaces of electrodes usually suffer from oxygen loss, transition metal dissolution and migration. Authors have reported that charged cathode electrode materials tend to react with the non-aqueous electrolyte violently at elevated temperatures or high voltage. Here we focus on the surface structure evolution of classical cathode materials, including layered, spinel, and olivine LiFePO 4 .
Layered cathode materials, such as LiCoO 2 , LiNi 1− x − y Co x Mn y O 2 (0 < x < 1, 0 < y < 1), LiNi 0.8 Co 0.15 Al 0.05 O 2 and lithium-rich manganese layered oxide are the most promising candidates for high energy-density lithium-ion batteries used in EVs or HEVs. A change in surface structure may destroy the two-dimensional Li + diffusion channels in these materials. Our recent investigation of well-crystallized LiCrO 2 cathode indicates that Cr ions can migrate into the Li layers in the surface regions, leading to an irreversible phase transformation from the layered to the rock-salt structure. The existence of the rock-salt phase in the surface blocks the extraction of lithium from the material. [ 27 ] Research has shown that during cycling, transition metal ions in both LiNi 1− x − y Co x Mn y O 2 and LiNi 0.8 Co 0.15 Al 0.05 O 2 migrate irreversibly and lead the surface structure to change from layered to spinel and rock-salt, resulting in capacity fade. [ 50 – 54 ] The oxygen loss from the surface of the material is the main reason. [ 54 ]
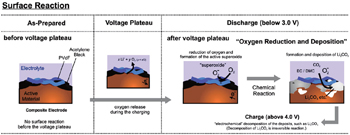
Lithium-rich manganese layered oxides materials, which have a high discharge capacity exceeding 250 mAh·g −1 , suffer many practical issues such as oxygen release, [ 55 ] voltage fade, [ 56 , 57 ] poor rate performance [ 58 ] and low coulomb efficiency. [ 59 – 61 ] Among these, oxygen release and voltage fade are believed to be related to changes of the materials’ surface structure. The high charge capacity of Li-rich layered cathode materials at high voltage arises mainly from the oxygen reduction reaction at the electrode surface. The proposed reaction schemes are summarized in Fig.
 | Fig. 3. Scheme of surface reaction mechanism in the Li-rich layered electrode. [ 62 ] Reprinted from Ref. [ 62 ]. Copyright 2011 American Chemical Society. |
The voltage fade mechanism is also related to the surface structure evolution during cycles. Zhang et al. systematically researched the phase transformation pathway in detail. [ 63 – 65 ] During the cycle, a phase transformation from the layered structure (initial C 2/ m phase transforms to R -3 m phase after activation) to a LT-LiCoO 2 type defect spinel-like structure (with the Fd -3 m space group) and then to a disordered rock-salt structure (with the Fm -3 m space group). The voltage fade can be well correlated with lithium-ion insertion into octahedral sites rather than tetrahedral sites, in both defect spinel-like and disordered rock-salt structures. Note that the transformation pathway of the Li-rich cathode is closely correlated to its initial structure and preparation conditions, and for a certain electrode, different direction may have different surface structure change. [ 53 , 66 , 67 ]
Surface dissolution of manganese is a long-standing issue for spinel cathode materials such as LiMn 2 O 4 and LiNi 0.5 Mn 1.5 O 4 . It is generally believed to be the main cause of their capacity fade, especially when cycling at high temperature. Our recent studies gained direct observation of the formation of a M 3 O 4 phase and rock-salt in spinel LiMn 2 O 4 and LiNi 0.5 Mn 1.5 O 4 . [ 20 , 68 ] Figure
 | Fig. 4. A typical STEM image and schematic lattice structures, showing the surface and subsurface regions of the initially charged LiNi 0.5 Mn 1.5 O 4 sample. [ 68 ] Reprinted from Ref. [ 68 ]. Copyright 2014 American Chemical Society. |
 | Fig. 5. Evolution of the chemical valence of Mn in electrodes based on LiNi 0.5 Mn 1.5 O 4 . (a) The electrodes with different SOC used for sXAS measurements are marked on the voltage profiles. (b) and (c) Mn L-edge sXAS spectra (TEY) collected on the sides of electrodes facing separator (b) and current collector (c). Dotted lines on top of the spectra are simulated results for obtaining the quantitative values of Mn 2+ , Mn 3+ , and Mn 4+ concentration. (d)–(f) Evolution of the Mn valence deduced from spectral simulations, with respect to the charge/discharge states. [ 69 ] |
LiFePO 4 is considered to have superior thermal stability, which is due to the strong P–O covalent bonds in the olivine structure. The stable structure can restrain oxygen release. LiFePO 4 in electrochemical cycling usually shows excellent stability. However at elevated temperatures, a performance fade can still be observed. [ 70 – 73 ] Electrochemical inactive LiFePO 4 (OH) will form at the surface of LiFePO 4 , when moisture (water) or hydroxyl groups contaminate the electrolytes. [ 43 ] This will lead the capacity fade and poor cycle performance.
These findings suggest that enhancing the stability of the surface structures of the electrode is the key to improve the long-term cycle performance of cathode materials. Efforts to stabilize the surface can significantly improve the capacity retention of cathodes. Representative approaches include different kinds of surface coating and electrolyte additives will be further discussed in the next two parts.
Forming stable SEI on cathode between the electrode and electrolyte is necessary to achieve satisfactory electrochemical performance of Li-ion batteries. [ 74 ] This interphase offers a channel for Li + , but it is insulated for electrons. It plays an important role in the irreversible capacity, cycle performance and stability. [ 75 ] Compared with the relatively clear understanding of the SEI on anodes, the knowledge of SEI on cathodes is quite insufficient. It is suggested that past studies, analysis and understanding of the SEI strongly underline the significance of its many aspects.
Three formation mechanisms are proposed for the SEI on cathodes: i) the SEI on the cathode is formed during the charge discharge process. [ 76 ] ii) the SEI is produced by reduction of the anode, and it diffuses and deposits on the cathode, changing the structure of the cathode. [ 74 ] iii) The SEI is a product of the chemical reaction between the electrode and electrolyte. [ 77 , 78 ] Wang et al. [ 36 , 79 , 80 ] found that LiCoO 2 spontaneously reacts with commercial electrolyte LiPF 6 /EC-DMC. FTIR and Raman spectrum show that the reduction products are (ROCO 2 Li) 2 and Li 2 CO 3 etc. Meanwhile, the surface of LiCoO 2 was reduced to Co 3 O 4 and Co 2 O 3 . Liu et al. [ 81 ] found that SEI can also form on the surface of nanosize LiCoO 2 particles after storage in dimethyl carbonate (DMC) solvent, and the SEI film becomes more uniform upon cycling. Figure
 | Fig. 6. TEM images of nanosize LiCoO 2 particle in different situations: (a) pristine, (b) soaked by 1 M LiPF 6 dissolved in EC/DMC (1:1 in volume) electrolyte for 7 days, (c) soaked by anhydrous DMC (< 10 ppm) for 7 days, and (d) after 10 cycles in a LiCoO 2 /Li cell at a voltage range of 2.5–4.3 V. [ 81 ] |
Some candidate additives have been reported to provide a stable SEI film for cathodes. Tris(pentafluorophenyl)borane ((C 6 F 5 ) 3 B, TPFPB) is an effective anion receptor. The released oxygen species in the form of oxygen anions 
Surface coating is the main method to improve the electrochemical performance and protect the cathode surface. The two most successful cases are carbon [ 91 ] and metal oxide [ 92 ] coating. The key mechanisms that have been proposed to explain the positive effect of surface coating on the performance of cathode materials include the following: [ 93 ] (i) electron-conducting media that facilitates the charge transfer at the surface of particles; (ii) modification of cathode surface chemistry that improves the cathode performance; (iii) HF scavenger that reduces the acidity of non-aqueous electrolyte and suppresses transition metal dissolution from the cathode materials; and (iv) a physical protection barrier that impedes the side reactions between cathode materials and non-aqueous electrolytes.
The major problem of olivine LiFePO 4 cathode material is its low electric conductivity and sluggish lithium diffusion. [ 94 ] This leads to the poor rate performance of uncoated LiFePO 4 . One of the main breakthroughs lies in the formation of a thin conductive carbon coating at the surface of LiFePO 4 nanoparticles. [ 91 ] Carbon coating can improve the electron transfer through the interface of the cathode material particles and provide extra electron-conducting pathways among the cathode material particles and between the cathode material particles and the current collector. The significant positive effect of carbon coating on the cathode performance has been widely reported. [ 95 , 96 ] Wang et al. [ 97 ] have reviewed the development of carbon coating on LiFePO 4 cathodes. However, the role of carbon is still not completely understood in spite of its importance in the rate capability and cycle stability of LiFePO 4 electrodes. [ 98 ]
No-carbon second phase (e.g. metal oxides, metal phosphate, and polymers) coating were also reported to modify the LiFePO 4 surface. [ 100 – 102 ] Cho et al. [ 103 ] reported an electrical network composed of a thin carbon layer coated on spinel LiMn 2 O 4 nanoclusters for ultrahigh-rate lithium-ion batteries. Carbon coating is also used in other cathode materials.
Cho et al. [ 92 , 104 ] first introduced the surface coating of ZrO 2 and Al 2 O 3 to LiCoO 2 layered materials. They pointed out that coating LiCoO 2 with oxides can improve the capacity retention of LiCoO 2 cycled to 4.4 V. A number of other groups have confirmed this finding. [ 48 , 49 ] In situ XRD studies by Wang et al. [ 16 , 105 , 106 ] provide that the contribution of the surface coating to the capacity retention is not due to suppression of the variation of lattice parameters but just the opposite: the variation of the lattice parameter is a structural change in response to the lithium intercalation/deintercalation. They also suggest that it is the surface of LiCoO 2 , not bulk structural changes, a phenomenon that holds the key to solving the problem of the capacity fade of LiCoO 2 during high-voltage cycling. Besides the metal oxide, AlF 3 , [ 107 – 109 ] AlPO 4 , [ 110 ] LiAlO 2 , [ 111 ] Li 3 PO 4 , [ 112 ] FePO 4 , [ 113 ] and MgO [ 114 ] etc. are also effective coating candidates. Using one active material to coat another active is also effective. [ 115 , 116 ]
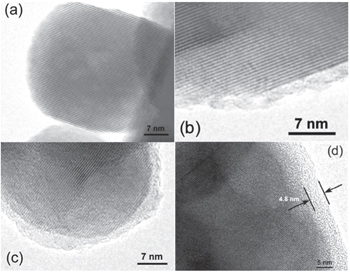
 | Fig. 7. (a) TEM image and (b) high-resolution TEM image of single-crystalline LiFePO 4 /C nanowires, [ 95 ] (c) TEM image and (d) high-resolution TEM image of LiFePO 4 /C nanoplate. [ 99 ] |
Recent years, ALD (atomic layer deposition) [ 113 , 117 – 120 ] is believed to be an important technique for the research of high performance cathode materials, for its advantage in excellent uniformity and conformity. Core–shell [ 121 – 124 ] and gradient materials [ 125 , 126 ] have been reported to combine the high electrochemical performance of the core material and the low reactivity of the shell material.
In the research of Li-ion batteries, it is important to know the bulk and surface structure evolution of the equilibrium and nonequilibrium states to understand the performance fade mechanism and improve it. Figure
 | Fig. 8. Space-resolution scheme of experimental techniques in Li-ion batteries. [ 127 ] |
In practical studies, it is usually necessary to combine two or more techniques to understand a scientific issue. For example, in the study on multi-electron transfer and structure evolution of Li 1.2 Cr 0.4 Mn 0.4 O 2 , h-XAS can give the average chemical state change that Cr 3+ are oxidized to Cr 6+ during charge, and migrated to tetrahedral sites in lithium layers; Mn 4+ is reduced to Mn 3+ during the discharge at low voltage. Cs-STEM can give the information that some transition metal at the surface has transferred to the lithium layer octahedral sites, forming ordered rock-salt structure. [ 31 ] S-XAS confirms that Mn 2+ may be contained at surface of the materials. Combining the tools for bulk and surface, the processes can be fully understood.
The behavior of cathodes for lithium ion batteries depends strongly on their surface chemistry. The surface chemistry stability is a big challenge during the cycle. In the past two decades, the energy density of Li-ion batteries (LiCoO 2 /C cell) has nearly reach the theoretical limits. The cycle performance has improved to over 800 cycles. In order to develop Li-ion batteries of adequate energy density and cycle performance, and excellent safety performance for EV and HEV, it is important to improve the surface stability of the cathode materials. Prevention of surface structural degradation can provide the means to produce and retain high capacity, as well as stabilize the cycle life of cathode materials for lithium ion batteries. Improving the reversibility of the oxygen redox reaction and restraining the dissolution and migration of transition metal ions at the electrode surface will result in the development of high-capacity cathode materials.
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 | |
| 22 | |
| 23 | |
| 24 | |
| 25 | |
| 26 | |
| 27 | |
| 28 | |
| 29 | |
| 30 | |
| 31 | |
| 32 | |
| 33 | |
| 34 | |
| 35 | |
| 36 | |
| 37 | |
| 38 | |
| 39 | |
| 40 | |
| 41 | |
| 42 | |
| 43 | |
| 44 | |
| 45 | |
| 46 | |
| 47 | |
| 48 | |
| 49 | |
| 50 | |
| 51 | |
| 52 | |
| 53 | |
| 54 | |
| 55 | |
| 56 | |
| 57 | |
| 58 | |
| 59 | |
| 60 | |
| 61 | |
| 62 | |
| 63 | |
| 64 | |
| 65 | |
| 66 | |
| 67 | |
| 68 | |
| 69 | |
| 70 | |
| 71 | |
| 72 | |
| 73 | |
| 74 | |
| 75 | |
| 76 | |
| 77 | |
| 78 | |
| 79 | |
| 80 | |
| 81 | |
| 82 | |
| 83 | |
| 84 | |
| 85 | |
| 86 | |
| 87 | |
| 88 | |
| 89 | |
| 90 | |
| 91 | |
| 92 | |
| 93 | |
| 94 | |
| 95 | |
| 96 | |
| 97 | |
| 98 | |
| 99 | |
| 100 | |
| 101 | |
| 102 | |
| 103 | |
| 104 | |
| 105 | |
| 106 | |
| 107 | |
| 108 | |
| 109 | |
| 110 | |
| 111 | |
| 112 | |
| 113 | |
| 114 | |
| 115 | |
| 116 | |
| 117 | |
| 118 | |
| 119 | |
| 120 | |
| 121 | |
| 122 | |
| 123 | |
| 124 | |
| 125 | |
| 126 | |
| 127 |