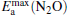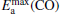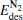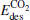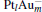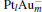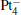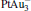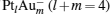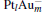1. IntroductionNitrous oxide (N2O), as a production from the combustion of fossil fuels, has been recognized as one of the contributors to global warming, and its global warming potential (GWP) is 300 times greater than that of carbon dioxide (CO2). N2O can also cause ozone depletion.[1] As another main harmful component in the stack gases, CO not only always co-exists with N2O in the combustion exhausts, but also is an excellent reducing gas. How to reduce the environmental pollution of N2O has been a hot subject for many experimental and theoretical research studies.[2– 9] The density functional theory (DFT) has been widely used to study the properties of molecules and materials, because DFT can reveal detailed mechanisms at atomic level for the cluster reactions.[10– 12]
The platinum-based catalyst is one of the most efficient catalysts for numerous important reactions, such as oxidation of CO to CO2.[13] The conversion of N2O and CO to N2 and CO2 catalyzed by neutral platinum clusters and their charged counterparts have been investigated.[14– 17] To the best of our knowledge, neutral platinum clusters and their positively charged counterparts are not active catalysts for the conversion of CO to CO2 with N2O. Fortunately, Siu et al. found that 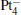 is a very active catalyst for the conversion of CO to CO2 with N2O.[18] However, the pure platinum catalyst is very expensive. So promising alternatives with high activity and lower price are required. Recently, the platinumbased bimetallic clusters have attracted special attention for their novel properties and high catalytic activities. It is proposed that the platinum catalysts alloyed or modified by other metals, such as ruthenium, palladium, and gold could exhibit higher activity for CO oxidation. Among the platinum-based bimetallic catalysts, platinum-gold nanoparticles are of particular interest for their superior catalytic performance.[19– 21] No study has been carried out involving the reduction of N2O by CO over platinum-gold bimetallic catalysts.
is a very active catalyst for the conversion of CO to CO2 with N2O.[18] However, the pure platinum catalyst is very expensive. So promising alternatives with high activity and lower price are required. Recently, the platinumbased bimetallic clusters have attracted special attention for their novel properties and high catalytic activities. It is proposed that the platinum catalysts alloyed or modified by other metals, such as ruthenium, palladium, and gold could exhibit higher activity for CO oxidation. Among the platinum-based bimetallic catalysts, platinum-gold nanoparticles are of particular interest for their superior catalytic performance.[19– 21] No study has been carried out involving the reduction of N2O by CO over platinum-gold bimetallic catalysts.
In this paper, we perform a density functional theory (DFT) study to investigate the catalytic properties of four negatively charged clusters 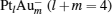 in the conversion of N2O and CO to N2 and CO2. We find that with the increase of the proportion of gold atoms in the cluster, the catalytic properties can be dramatically enhanced, and as the percentage of platinum is reduced, the cost of the catalyst is greatly reduced.
in the conversion of N2O and CO to N2 and CO2. We find that with the increase of the proportion of gold atoms in the cluster, the catalytic properties can be dramatically enhanced, and as the percentage of platinum is reduced, the cost of the catalyst is greatly reduced.
2. CalculationsAll calculations were carried out using the density functional theory with the generalized gradient approximation (GGA) and the revised Perdew, Burke, and Ernzerhof exchange– correlation (RPBE)[22] as implemented in the DMOL3 computational software package.[23, 24] The strong relativistic effect of Pt and Au has been considered. The spin polarization was included and no symmetric constraints were imposed in all calculations. In the self-consistent-field (SCF) calculation, the optimization convergence thresholds for energy change, maximum force, and maximum displacement were 2.0 × 10− 5 Ha, 4.0 × 10− 3 Ha/Å , and 5.0 × 10− 3 Å , respectively.
The overall reduction of N2O by CO can be written as
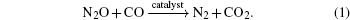
It can be divided into two elementary reactions
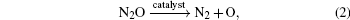
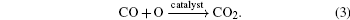
The adsorption energy Eads of N2O to 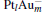 clusters is defined as
clusters is defined as
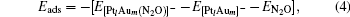
where E[PtlAum(N2O)]− is the total energy of the absorption system, E[PtlAum]− is the total energy of the 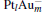 clusters, and EN2O is the total energy of an isolated N2O molecule.
clusters, and EN2O is the total energy of an isolated N2O molecule.
The desorption energy 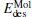 of N2 or CO2 from the cluster can be obtained with the following equation:
of N2 or CO2 from the cluster can be obtained with the following equation:
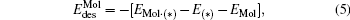
where EMol· (* ) is the total energy of the [PtlAumO· N2]− or [PtlAum· CO2]− cluster; E(* ) is the total energy of the [PtlAumO]− or [PtlAum]− cluster; and EMol is the total energy of an isolated N2 or CO2 molecule.
The dissociation energy Edis for the negatively charged 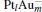 clusters is defined as
clusters is defined as
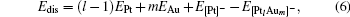
where EPt, EAu, and E[Pt]− are the total energies of the Pt, Au, and Pt− atoms, respectively.
In order to analyze the catalytic activity of the 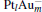 clusters with different weights of Au, we use the average energy of the occupied M 5d band center (Ed), which is given by
clusters with different weights of Au, we use the average energy of the occupied M 5d band center (Ed), which is given by
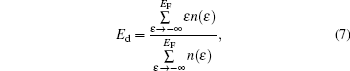
where EF is the Fermi level, and n(ɛ ) is the Mulliken population of the energy level ɛ for M 5d orbitals. The occupied M 5d band center is marked by the black arrows in Fig. 3.
The activation energies for the above two elementary reactions are identified by complete linear synchronous transition and quadratic synchronous transit search methods, following by transition-state optimization and confirmation. The optimized transitional points on the potential energy surface are verified by the second-order derivatives of the energy with respect to the atomic coordinates (Hessian) through vibrational frequency calculations.
3. Results and discussion3.1. Binding and dissociation of N2O on 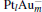 cluster
clusterIn Table 1, we list the adsorption energy of N2O (Eads), the largest activate energies 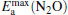 (for the elementary reaction N2O → N2 + O) and
(for the elementary reaction N2O → N2 + O) and 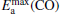 (for the elementary reaction CO + O → CO2), and the desorption energies
(for the elementary reaction CO + O → CO2), and the desorption energies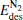 (for N2) and
(for N2) and 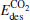 (for CO2) in the reaction pathway of the
(for CO2) in the reaction pathway of the 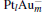 clusters. The dissociation energy Edis of the
clusters. The dissociation energy Edis of the 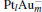 clusters is also listed (the value in the parenthesis is Edis of the PtlAum clusters). We find that Edis for the negatively charged
clusters is also listed (the value in the parenthesis is Edis of the PtlAum clusters). We find that Edis for the negatively charged 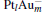 clusters is larger than that for their neutral counterparts. At the same time, all Edis for the
clusters is larger than that for their neutral counterparts. At the same time, all Edis for the 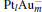 clusters are larger than 2.00 eV. It means that the
clusters are larger than 2.00 eV. It means that the 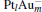 clusters are more stable than their neutral counterparts.
clusters are more stable than their neutral counterparts.
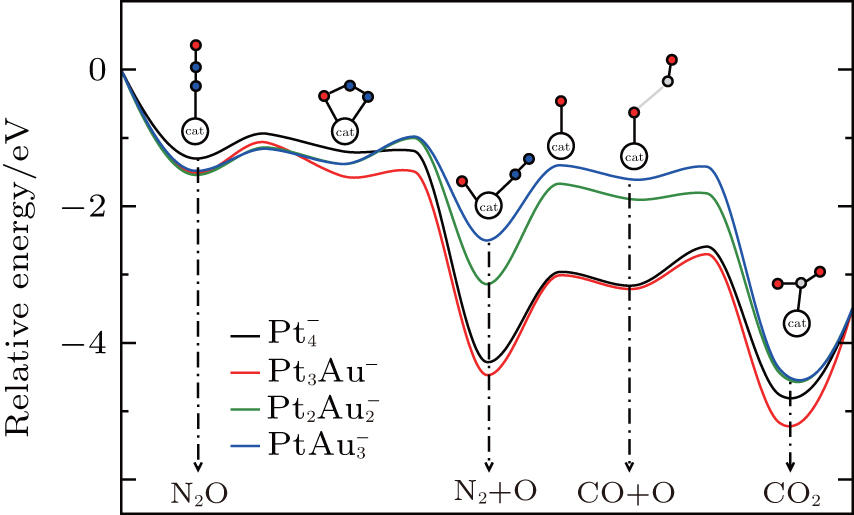
Figure 1 shows the most stable structures and their frontier molecular orbitals (FMOs) isosurfaces for CO, N2O, 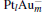 , and [PtlAum(N2O)]− clusters. N2O has a linear structure with C∞ v point symmetry. The bond lengths of N– N and N– O are 1.13 Å and 1.20 Å in this work, which are in very good agreement with the experimental results of 1.14 Å and 1.18 Å , respectively. The reduction of N2O by CO in the absence of a catalyst has a high activation barrier of about 47.2 kcal/mol, and is exothermic by 87.0 kcal/mol.[9] So the binding of N2O molecules on catalysts is considered as a necessary step for the consequent N2O decomposition.
, and [PtlAum(N2O)]− clusters. N2O has a linear structure with C∞ v point symmetry. The bond lengths of N– N and N– O are 1.13 Å and 1.20 Å in this work, which are in very good agreement with the experimental results of 1.14 Å and 1.18 Å , respectively. The reduction of N2O by CO in the absence of a catalyst has a high activation barrier of about 47.2 kcal/mol, and is exothermic by 87.0 kcal/mol.[9] So the binding of N2O molecules on catalysts is considered as a necessary step for the consequent N2O decomposition.
To find the most favorable geometries of the [PtlAum(N2O)]− clusters, various possible binding sites of the molecule on the clusters and different relative orientations between the cluster and the molecule have been used as the initial geometries. The adsorption site of the molecule on the catalyst can be predicted with the frontier molecular orbital theory by observing the interaction between the highest occupied orbital (HOMO) and the lowest unoccupied molecule orbital (LUMO).[25]
We take 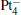 as an example to illustrate how it works. The
as an example to illustrate how it works. The 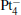 cluster has a rhombus structure as shown in Fig. 1. The HOMO and LUMO of
cluster has a rhombus structure as shown in Fig. 1. The HOMO and LUMO of 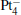 mainly locate at the Pt atom of the inner obtuse angle, while the HOMO and LUMO of the N2O molecule mainly locate at the terminal N atom. When the N2O molecule is imported, the terminal N atom is bound to the Pt atom of the inner obtuse angle to constitute the [Pt4(N2O)]− cluster. Our DFT calculations confirm this result. Incidentally, with more Pt atoms replaced by Au atoms, the interaction between N2O and the catalyst drastically modifies the structure of the cluster, as shown in Fig. 1 for Pt3Au− ,
mainly locate at the Pt atom of the inner obtuse angle, while the HOMO and LUMO of the N2O molecule mainly locate at the terminal N atom. When the N2O molecule is imported, the terminal N atom is bound to the Pt atom of the inner obtuse angle to constitute the [Pt4(N2O)]− cluster. Our DFT calculations confirm this result. Incidentally, with more Pt atoms replaced by Au atoms, the interaction between N2O and the catalyst drastically modifies the structure of the cluster, as shown in Fig. 1 for Pt3Au− , 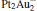 , and
, and 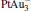 . It may be caused by the net charge transfer from the metal cluster to the N2O molecule when the N2O molecule is adsorbed on the
. It may be caused by the net charge transfer from the metal cluster to the N2O molecule when the N2O molecule is adsorbed on the 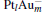 cluster. For example, without adsorbing N2O, the structure of [PtAu3]− is a caped triangle; it changes to the rhombus structure when N2O is adsorbed.
cluster. For example, without adsorbing N2O, the structure of [PtAu3]− is a caped triangle; it changes to the rhombus structure when N2O is adsorbed.
When the N2O molecule is adsorbed on the 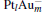 cluster, the N– O bond length of the N2O molecule is significantly elongated compared to its gas-phase value (1.20 Å ). Generally, the N– O bond length is around 1.24 Å for all four
cluster, the N– O bond length of the N2O molecule is significantly elongated compared to its gas-phase value (1.20 Å ). Generally, the N– O bond length is around 1.24 Å for all four 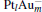 clusters. In our study, when N2O is bound to the clusters, the calculated Eads of N2O on Pt site in Pt3Au− ,
clusters. In our study, when N2O is bound to the clusters, the calculated Eads of N2O on Pt site in Pt3Au− , 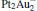 ,
, 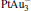 are 1.51 eV, 1.54 eV, and 1.49 eV (Table 1), respectively. These values are slightly larger than the corresponding adsorption energy on the
are 1.51 eV, 1.54 eV, and 1.49 eV (Table 1), respectively. These values are slightly larger than the corresponding adsorption energy on the 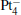 cluster (1.30 eV).
cluster (1.30 eV).
3.2. Reaction mechanism of N2O with CO over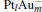 cluster
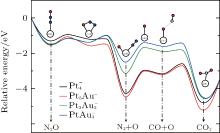
clusterFigure 2 represents the potential energy surface profiles for the reduction of N2O by CO over the 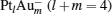 catalysts. The energies of the intermediates (IM), the transition states (TS), and the products are referred to the energy of the entrance (the free catalyst, one isolated N2O molecule, and one isolated CO molecule). The catalytic reaction pathways over the
catalysts. The energies of the intermediates (IM), the transition states (TS), and the products are referred to the energy of the entrance (the free catalyst, one isolated N2O molecule, and one isolated CO molecule). The catalytic reaction pathways over the 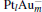 clusters, with the intermediates and the transition states, are also shown in Fig. 2.
clusters, with the intermediates and the transition states, are also shown in Fig. 2.
From Fig. 2, it can be concluded that the reaction processes over the 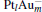 catalysts are as follows. Firstly, a N2O molecule is adsorbed on the optimized catalyst, forming a bridged intermediate state, and then the N– O bond is broken, a N2 molecule forms and desorbs, which generates a corresponding oxidized cluster. Secondly, the oxygen atom of the oxidized cluster is seized by a CO molecule and one CO2 molecule forms, then the CO2 is desorbed by the catalyst.
catalysts are as follows. Firstly, a N2O molecule is adsorbed on the optimized catalyst, forming a bridged intermediate state, and then the N– O bond is broken, a N2 molecule forms and desorbs, which generates a corresponding oxidized cluster. Secondly, the oxygen atom of the oxidized cluster is seized by a CO molecule and one CO2 molecule forms, then the CO2 is desorbed by the catalyst.
We now illustrate the catalytic reaction over the Pt3Au− cluster. The N2O molecule adsorbed on the Pt3Au− catalyst forms intermediate IM1 [Pt3Au(N2O)]− , which locates at 1.51 eV below the entrance. The oxygen atom in the N2O molecule is activated and the bond length of N– O is elongated (to 1.24 Å ). Then the adsorbed N2O bends toward an adjacent Pt atom and forms a bridged intermediate IM2 via transition state TS12 with a barrier of 0.44 eV. IM2 converts into IM3 and forms compound [OPt3AuN2]− via transition state TS23 with a barrier of 0.06 eV, in which the N– O bond is broken. This step is strongly exothermic (4.47 eV). After the N2 is desorbed, the oxygen atom in the N2O molecule is combined to the Pt3Au− cluster, forming IM4, which is an oxidized cluster. Now a CO molecule is imported to bind with the oxygen atom, forming the intermediate IM5, which locates at − 3.21 eV below the entrance. IM5 converts to IM6 via transition state TS56 with a barrier of 0.51 eV. IM5 dissociates directly to the CO2 molecule and the Pt3Au− cluster, indicating the accomplishment of the reaction.
As shown in Table 1, for the 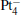 cluster,
cluster, 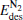 (1.32 eV) is a little larger than Eads (1.30 eV), at the same time,
(1.32 eV) is a little larger than Eads (1.30 eV), at the same time, 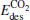 (1.28 eV) is a little smaller than Eads (1.30 eV). With one Pt atom replaced by an Au atom in the
(1.28 eV) is a little smaller than Eads (1.30 eV). With one Pt atom replaced by an Au atom in the 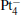 cluster, the desorption energy
cluster, the desorption energy 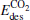 (1.69 eV) is larger than Eads (1.51 eV) for the Pt3Au− cluster, which indicates that the catalyst is degraded. However, with more Pt atoms replaced by Au atoms, the desorption energies
(1.69 eV) is larger than Eads (1.51 eV) for the Pt3Au− cluster, which indicates that the catalyst is degraded. However, with more Pt atoms replaced by Au atoms, the desorption energies 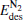 and
and 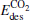 both decrease remarkably. The Eads is larger than the desorption energies
both decrease remarkably. The Eads is larger than the desorption energies 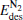 and
and 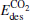 meanwhile
meanwhile 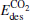 is smaller than
is smaller than 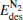 , which means that the poisoning is overcome. From Table 1, we also find that, as the proportion of Au atoms in the
, which means that the poisoning is overcome. From Table 1, we also find that, as the proportion of Au atoms in the 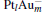 clusters increases, the activate energy
clusters increases, the activate energy 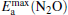 for the elementary reaction N2O → N2 + O decreases from 0.44 eV to 0.39 eV, and
for the elementary reaction N2O → N2 + O decreases from 0.44 eV to 0.39 eV, and 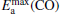 for the elementary reaction CO + O → CO2 decreases remarkably. It means that with more Pt atoms replaced by Au atoms, more N2O molecules are dissociated and more CO molecules are oxidized. So the catalytic performance can be improved via the replacement of more Pt atoms by Au atoms in the negatively charged cluster
for the elementary reaction CO + O → CO2 decreases remarkably. It means that with more Pt atoms replaced by Au atoms, more N2O molecules are dissociated and more CO molecules are oxidized. So the catalytic performance can be improved via the replacement of more Pt atoms by Au atoms in the negatively charged cluster 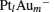 cluster.
cluster.
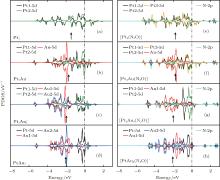
3.3. Electronic properties of the catalystsThe projected densities of states (PDOS) of M 5d (metal 5d) and N 2p orbits for 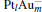 (left panel) and [PtlAum(N2O)]− (right panel) are shown in Fig. 3. The occupied M 5d band centers are also indicated by the black arrows. The occupation numbers of the 5d6s6p orbitals in each atom for the
(left panel) and [PtlAum(N2O)]− (right panel) are shown in Fig. 3. The occupied M 5d band centers are also indicated by the black arrows. The occupation numbers of the 5d6s6p orbitals in each atom for the 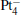 ,
, 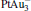 [Pt4(N2O)]− , and [PtAu3(N2O)]− clusters are shown in Table 2. Without the adsorption of N2O to the
[Pt4(N2O)]− , and [PtAu3(N2O)]− clusters are shown in Table 2. Without the adsorption of N2O to the 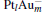 clusters, there are some peaks in the PDOS of the M 5d over the Fermi level (as shown in the left panel of Fig. 3). When the N2O molecule is adsorbed on the
clusters, there are some peaks in the PDOS of the M 5d over the Fermi level (as shown in the left panel of Fig. 3). When the N2O molecule is adsorbed on the 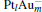 clusters, the peaks for the majority M 5d states disappear, as shown in the right panel of Fig. 3. At the same time, the PDOS of the N 2p emerges on both sides far away from the Fermi level. In Table 2, we also notice that the occupation number for the Pt(1) atom changes from 5d8.796s1.016p0.31 to 5d8.606s0.926p0.41 when the N2O molecule is bound to the Pt(1) atom (
clusters, the peaks for the majority M 5d states disappear, as shown in the right panel of Fig. 3. At the same time, the PDOS of the N 2p emerges on both sides far away from the Fermi level. In Table 2, we also notice that the occupation number for the Pt(1) atom changes from 5d8.796s1.016p0.31 to 5d8.606s0.926p0.41 when the N2O molecule is bound to the Pt(1) atom (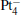 to [Pt4(N2O)]− ). For
to [Pt4(N2O)]− ). For 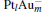 and [PtlAum(N2O)]− , we find that the changes of the occupation numbers on 5d6s6p for the Pt atom adsorbed with N atom are similar. These clues indicate that one of the unoccupied M 6p orbitals interacts with the lone electron pair (LEP) of N2O to form the σ coordination bond, while the partial bonding 5d orbitals and the anti-bonding π * orbital of N2O form the back donation π coordination bond. The σ and π coordination bonds make the compound more stable, which leads to the larger Eads for the
and [PtlAum(N2O)]− , we find that the changes of the occupation numbers on 5d6s6p for the Pt atom adsorbed with N atom are similar. These clues indicate that one of the unoccupied M 6p orbitals interacts with the lone electron pair (LEP) of N2O to form the σ coordination bond, while the partial bonding 5d orbitals and the anti-bonding π * orbital of N2O form the back donation π coordination bond. The σ and π coordination bonds make the compound more stable, which leads to the larger Eads for the 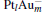 clusters (as shown in Table 1). The larger Eads will enhance the catalytic performance.
clusters (as shown in Table 1). The larger Eads will enhance the catalytic performance.
From the left panel of Fig. 3, it can be seen that, with increasing proportion of Au in the 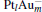 clusters, the width of PDOS for M 5d declines, and the PDOS of M 5d is concentrated on one area from (− 5.5 eV, 1.0 eV) to (− 3.8 eV, 0.2 eV), which means that the catalytic activity of more inner M-5d electron will be improved. At the same time, there is strong orbital hybridization between the Pt and Au atoms, which results in the down shift of the M-5d band center of the
clusters, the width of PDOS for M 5d declines, and the PDOS of M 5d is concentrated on one area from (− 5.5 eV, 1.0 eV) to (− 3.8 eV, 0.2 eV), which means that the catalytic activity of more inner M-5d electron will be improved. At the same time, there is strong orbital hybridization between the Pt and Au atoms, which results in the down shift of the M-5d band center of the 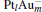 clusters away from the Fermi level. It is interesting to find that, as the M-5d band center of
clusters away from the Fermi level. It is interesting to find that, as the M-5d band center of 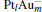 shifts down (shown in the left panel of Fig. 3), the largest activate energies for the elementary reaction N2O → N2 + O and the elementary reaction CO + O → CO2 are decreased remarkably (shown in Fig. 2 and Table 1), at the same time, the desorption energies
shifts down (shown in the left panel of Fig. 3), the largest activate energies for the elementary reaction N2O → N2 + O and the elementary reaction CO + O → CO2 are decreased remarkably (shown in Fig. 2 and Table 1), at the same time, the desorption energies 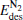 and
and 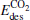 in the reaction pathway (shown in Fig. 2 and Table 1) are reduced remarkably. This is consistent with the improved catalytic properties of
in the reaction pathway (shown in Fig. 2 and Table 1) are reduced remarkably. This is consistent with the improved catalytic properties of 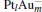 with increasing proportion of Au in the bimetallic cluster. So the catalytic activity will be enhanced by the synergetic effect between Pt and Au atoms in bimetallic clusters
with increasing proportion of Au in the bimetallic cluster. So the catalytic activity will be enhanced by the synergetic effect between Pt and Au atoms in bimetallic clusters 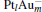 compared to pure
compared to pure 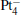 or
or 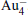 .
.
It is interesting to find that the reaction center is the Pt atom as shown in Fig. 1. When the N2O molecule is adsorbed on the  clusters, the terminal N atom is bound to the Pt atom. If the N2O molecule is bound to the Au atom, the adsorption energy Eads is very small, which means that the interaction between N2O and the metal cluster is very weak, then the first step of the catalytic reaction cannot proceed. This is why
clusters, the terminal N atom is bound to the Pt atom. If the N2O molecule is bound to the Au atom, the adsorption energy Eads is very small, which means that the interaction between N2O and the metal cluster is very weak, then the first step of the catalytic reaction cannot proceed. This is why  shows little catalytic activity for the reaction of N2O and CO. We also notice that the σ – π coordination bonds enhance the interaction between the metal cluster and N2O, and result in a net charge transfer from the metal cluster to the N2O molecule, which weakens the N– O bond in the N2O molecule and results in longer N– O bond. Therefore, the N2O molecule is bent to the metal cluster and the N– O bond is easily broken. So the Pt atom in the
shows little catalytic activity for the reaction of N2O and CO. We also notice that the σ – π coordination bonds enhance the interaction between the metal cluster and N2O, and result in a net charge transfer from the metal cluster to the N2O molecule, which weakens the N– O bond in the N2O molecule and results in longer N– O bond. Therefore, the N2O molecule is bent to the metal cluster and the N– O bond is easily broken. So the Pt atom in the 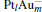 cluster is the reaction center, and all metal atoms act as electron donors.
cluster is the reaction center, and all metal atoms act as electron donors.
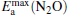
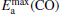
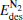
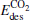
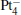
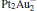
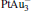
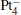
{kind=link}
{kind=link}
{kind=link}
 , Duan Xiang-Mei
, Duan Xiang-Mei