





{kind=link}
{kind=link}
Elastic properties and electronic structures of lanthanide hexaborides
[Duan Jiea) , Zhou Tonga) , Zhang Li†a)  , Du Ji-Guang
, Du Ji-Guangb) , Jiang Ganga) , Wang Hong-Bina) ]
, Du Ji-Guang|
|


†Corresponding author. E-mail: lizhang@scu.edu.cn
The structural, elastic, and electronic properties of a series of lanthanide hexaborides ( LnB6) have been investigated by performing ab initio calculations based on the density functional theory using the Vienna ab initio simulation package. The calculated lattice and elastic constants of LnB6 are in good agreement with the available experimental data and other theoretical results. The polycrystalline Young’s modulus, shear modulus, the ratio of bulk to shear modulus B/G, Poisson’s ratios, Zener anisotropy factors, as well as the Debye temperature are calculated, and all of the properties display some regularity with increasing atomic number of lanthanide atoms, whereas anomalies are observed for EuB6 and YbB6. In addition, detailed electronic structure calculations are carried out to shed light on the peculiar elastic properties of LnB6. The total density of states demonstrates the existence of a pseudogap and indicates lower structure stability of EuB6 and YbB6 compared with others.
Lanthanide hexaborides (LnB6: Ln = La, Ce, Pr, Nd, Pm, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb, Lu) have attracted extensive experimental and theoretical interest due to their intriguing physical properties. For example, LaB6 is a typical metal and becomes superconducting at TC = 0.45 K, [1, 2] CeB6 is a dense Kondo material and shows heavy fermion behavior.[3] SmB6, a typical mixed valence compound, has been theoretically proven to be a topological Kondo insulator, which is supported by recent photo emission and transport experiments.[4, 5] YbB6 is found to be a moderately correlated Z2 topological insulator, similar to SmB6 but having a much larger bulk band gap.[6] Furthermore, the lanthanide hexaborides are also considered as hard and refractory materials because of their special cage structure.[7] Bulk lanthanide hexaborides have low work functions ranging from 2.1 eV to 3.8 eV, which make them excellent candidate materials for electron emitter devices.[8] Moreover, they have been applied as scratch-resistant surface decorative coatings due to their tunable attractive colors. For instance, the stoichiometric LaB6 is usually purple, while its color turns to deep blue when it is lanthanum deficient. Consequently, the investigation of the basis physical properties of a series of lanthanide hexaborides such as structural, elastic, and electronic properties is necessary and offers credible references for their various applications.
In general, the elastic properties of a solid are important because some physical properties, such as the bulk modulus, shear modulus, Young’ s modulus, and Poisson’ s ratio can be derived from the elastic constants.[9] Up to now, most studies have focused on the magnetic and thermodynamic properties of LnB6.[10] The structural and elastic properties of some LnB6 have also been investigated experimentally by several groups but their results are relatively contradictory or insufficient. Some significantly different values have been reported for the measured elastic constants of CeB6, especially C12; Takegahara et al.[11] gave C12 = − 93 GPa while Nakamura et al.[12] reported a value of 53 GPa for the same constant. The bulk moduli also show small differences among the various experimental reports.[13] On the other hand, single crystal elastic constants for the LnB6 compounds are scarce due to the difficulties of the experiments. The elastic properties of a solid are also closely associated with various fundamental solid-state properties, such as phonon spectra, specific heat, Debye temperature, etc. In view of this, it is necessary and essential to obtain their elastic constants by the first-principles calculations.
As we all know, in the lanthanide series both Eu and Yb are divalent in the solid state while all of the others are in the trivalent state. There is a substantial energy penalty for Eu and Yb to be in the trivalent state. Consequently, systematics are observed for the trivalent Ln and their compounds in terms of crystal structures and physical properties, whereas anomalies are observed for Eu and Yb.[14] The abnormalities relating to Eu and Yb are probably due to their special electronic configurations. Eu has a half-filled 4f orbital and Yb has a completely filled 4f orbital; therefore, they have stable configurations of low energy. But their compounds disturb the stable electronic configurations through charge transfer or chemical bonding. The deviations will be seen repeatedly throughout this work.
The main objective of this paper is to study the regularity of elastic properties and electronic structure of the LnB6 compounds. This paper is organized as follows. The computational methodologies employed in the current study are given in Section 2. We present and discuss the results obtained and compare them with the available experimental and theoretical data in Section 3. Finally, the results are summarized in Section 4.
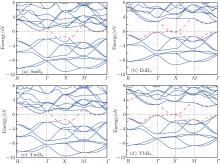
Our first-principles electronic structure calculations are based on the density functional theory (DFT) in conjunction with projector augmented wave potentials (PAW)[15, 16] within the generalized gradient approximations (GGA) of Perdew– Burke– Ernzerhof (PBE) as implemented in the Vienna ab initio simulation package (VASP).[17– 20] Generally, the PAW potentials are more reliable than the ultra-soft pseudopotentials because of the smaller cut off radius and the more accurate valence electron wave functions in the nuclear area. The validity for the LnB6 binary compounds has been widely verified.[12] Two choices of potentials are available for each Ln element: a standard version in which the entire set of f levels are treated within the valence band and a divalent or trivalent version (e.g., Yb2+ for Yb and Pm3+ for Pm) in which some f electrons are kept frozen in the core. There are several exceptions: (i) there is only a standard potential available for La because it has no occupied f levels in its elemental state, and (ii) there is only a trivalent version of potential available for Tb, Dy, Ho, and Er with VASP. Furthermore, the previous studies of the ground state structures, formation energies, and elastic constants of the Ln elements and compounds[21] indicate that the f core approach is the correct method to treat the Ln elements when thermodynamic and elastic properties are of interest. In this work, pseudopotentials with the f electrons frozen in the core were used throughout. A plane-wave energy cutoff of 550 eV was taken for all LnB6. The 13× 13× 13 k-points for LnB6 unit cell with the Monkhorst– Pack scheme were adopted for the Brillouin zone sampling. In all cases, the total energy of self-consistent convergence was 10− 5 eV/cell and the process was terminated when the atomic force was less than 10− 3 eV/Å . Spin polarization with ferromagnetic ordering was used in all calculations and we found that all the LnB6 considered here are not magnetic. To produce more accurate densities of states, a dense k-point mesh of 19 × 19 × 19 was used, and the total DOS was computed by the Gaussian smearing method.[17– 20] The chosen plane-wave cut off and numbers of k points have been tested carefully to ensure good converged results for all of the computations. The elastic constants were calculated through the energy strain method. The analysis of the density of states indicated the existence of a pseudogap.
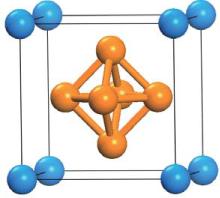
As illustrated in Fig. 1, the lanthanide hexaborides crystallize in cubic CsCl-type structure which belongs to the space group 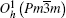
 | Fig. 1. Unit cell of lanthanide hexaboride (LnB6). |
 | Fig. 2. The change of lattice parameter a0 with the atomic number of Ln atoms in LnB6. |
Table 1. Calculated lattice parameters a0, positional parameter of boron atoms x, bulk modulus B0, equilibrium volume V, pressure derivative  |
The lattice constants, elastic properties, and electronic structures of LnB6 (Ln = La, Ce, … , Yb, Lu) have been studied comprehensively by the PAW method within the GGA. The ab initio calculated lattice constants and the elastic properties are in good agreement with the experimental data and other theoretical results available.
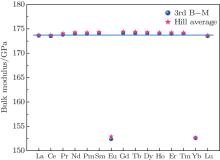
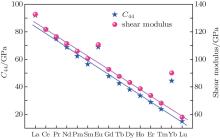
The lattice constants are compressed from LaB6 to LuB6 due to the famous lanthanide contraction effect. The independent elastic constants are investigated and the bulk moduli, shear moduli, Young’ s moduli, Poisson’ s ratios, and Zener anisotropy factors are also estimated for the LnB6 polycrystals. Our results show that the change of the shear modulus is similar to that of C44 and the hardness reduces gradually from LaB6 to LuB6. The analysis of the electronic density of states indicates the presence of a pseudogap. The total densities of states of EuB6 and YbB6 shift towards higher energy, which is inconsistent with their lower bulk moduli. We predict the Debye temperatures for LnB6 and they gradually decrease except for EuB6 and YbB6. The compounds of the divalent Ln elements Eu and Yb are usually exceptions to the trends, which holds mainly for the adoption of their divalent configurations due to their special electron configurations. Since there are no experimental data available for some of these parameters, we believe that our calculated results also provide a reference for future experimental work.
| 1 |
|
| 2 |
|
| 3 |
|
| 4 |
|
| 5 |
|
| 6 |
|
| 7 |
|
| 8 |
|
| 9 |
|
| 10 |
|
| 11 |
|
| 12 |
|
| 13 |
|
| 14 |
|
| 15 |
|
| 16 |
|
| 17 |
|
| 18 |
|
| 19 |
|
| 20 |
|
| 21 |
|
| 22 |
|
| 23 |
|
| 24 |
|
| 25 |
|
| 26 |
|
| 27 |
|
| 28 |
|
| 29 |
|
| 30 |
|
| 31 |
|
| 32 |
|
| 33 |
|
| 34 |
|
| 35 |
|
| 36 |
|
| 37 |
|
| 38 |
|
| 39 |
|
| 40 |
|
| 41 |
|
| 42 |
|
| 43 |
|
| 44 |
|
| 45 |
|
| 46 |
|
| 47 |
|