{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Stability of conductance oscillations in carbon atomic chains*
[Yu Jing-Xin† , Hou Zhi-Wei, Liu Xiu-Ying]
, Hou Zhi-Wei, Liu Xiu-Ying]
, Hou Zhi-Wei, Liu Xiu-Ying]
|
|


†Corresponding author. E-mail: j.x.yu@qq.com
*Project supported by the National Natural Science Foundation of China (Grant Nos. 11304079, 11404094, and 51201059), the Priority Scientific and Technological Project of Henan Province, China (Grant No. 14A140027), the School Fund (Grant No. 2012BS055), and the Plan of Natural Science Fundamental Research of Henan University of Technology, China (Grant No. 2014JCYJ15).
The conductance stabilities of carbon atomic chains (CACs) with different lengths are investigated by performing theoretical calculations using the nonequilibrium Green’s function method combined with density functional theory. Regular even–odd conductance oscillation is observed as a function of the wire length. This oscillation is influenced delicately by changes in the end carbon or sulfur atoms as well as variations in coupling strength between the chain and leads. The lowest unoccupied molecular orbital in odd-numbered chains is the main transmission channel, whereas the conductance remains relatively small for even-numbered chains and a significant drift in the highest occupied molecular orbital resonance toward higher energies is observed as the number of carbon atoms increases. The amplitude of the conductance oscillation is predicted to be relatively stable based on a thiol joint between the chain and leads. Results show that the current–voltage evolution of CACs can be affected by the chain length. The differential and second derivatives of the conductance are also provided.
Carbon atomic chains (CACs) have long been advocated as ideal one-dimensional (1D) molecular wires.[1, 2] These chains have attracted much attention in recent decades as an allotrope of carbon because of their applications as an emitting structure of atomic-scale field emitters[3] and atomic-scale electronic devices, [4, 5] However, the synthesis of high-quality CACs is difficult. Several experimental methods, such as chemical synthesis, [6]β -SiC surface-supported formation, and the double-anode arc-discharge method, [7] have been used to form CACs. Chains of up to 20 carbon atoms have been attached to metal atoms.[8] Long, stable, and free-standing linear atomic carbon wires (carbon chains) were recently carved out from grapheme using a high-energy electron beam.[9, 10] Chalifoux and Tykwinski also synthesized the longest polyyne — a linear chain of carbon atoms reported thus far by constructing a polyyne consisting of 44 carbon atoms; prior to this work, the longest chain reported only had 28 atoms.[11]
Many interesting physical properties of CACs have been explained theoretically, including nanoelectronic/spintronic devices.[12– 17] The electronic transport properties of a nanostructure are usually at the heart of the nanostructure’ s own functionality.[18] The sp bonding interface (carbon wire-carbon wire junctions), or the sp2 bonding interface (carbon wire-graphene junctions), or the sp3 bonding interface (carbon wire-metal and carbon wire-carbon nanotube junctions) shows several unique electron transport properties.[19] Lang and Avouries[5, 20] reported that the conductance of carbon chains is always oscillatory with even-numbered chains lower than odd-numbered ones. Experiments have shown that atomic carbon linear chains directly linked to two graphene ribbons can be formed by electron bombardment of graphene sheets.[11] Many investigations have focused on the electron transport properties of atomic nanowires between grapheme electrodes.[19, 21– 23] Shen et al.[19] showed that linear ballistic transport in short carbon chains may be observed in wires consisting of odd-numbered carbon atoms but not in those comprised of even-numbered carbon atoms. Larade et al.[24] reported a negative differential conductance when the carbon chain directly contacts Al nanowires. The current– voltage (I– V) curves exhibit two plateaus in even-numbered chains attached to Au electrodes through sulfur.[25] When capped with benzene-thiol attached to gold electrodes with amino and nitro groups, however, these chains show similar I– V characteristics, and the transport properties are almost independent of the molecular length.[26] Spin transport is also feasible in the presence of weak spin– orbit coupling in carbon atoms, and the spin relaxation lifetime is enhanced by gold deposition onto graphene sheets.[27] Carbyne, an infinite chain of sp-hybridized carbon atoms, was recently theoretically confirmed to have an extreme tensile stiffness surpassing that of any other known material.[28] In the future design of molecular electronic circuits, the nice control of the contact geometry is a must. Although a considerable body of literature on CAC electronic transport properties exists, providing as many configurations as possible by calculating different connections between the molecule and electrodes is necessary, considering that CAC is a fundamental form of many allotropes.
This article presents the results of theoretical studies on the electronic transport properties of CACs capped with carbon or sulfur atoms. The number of atoms in the carbon chain varies from 1 to 12. Even– odd conductance oscillation is influenced by changes in end atoms and variations in coupling strength between the chain and leads. I– V evolution is affected by the chain length when chain ends are capped with carbon or sulfur atoms.
First-principles simulations were performed for CACs or CACs capped with a sulfur atom sandwiched between semi-infinite Au electrodes with (100) orientation to elucidate the electronic structures and transport properties of CACs. The structures and properties were calculated using the ab initio transport code SMEAGOL, [29– 31] which is a fully self-consistent method that combines the non-equilibrium Green function (NEGF)[32, 33] with density functional theory (DFT).[34] SMEAGOL utilized the single-particle Kohn– Sham Hamiltonian provided by the DFT code SIESTA.[35] Zero-bias electron transmission coefficients and the I– V curves of two-terminal devices are calculated using this method. SMEAGOL can carry out fully self-consistent calculations of the electrical properties of devices formed by atomic-scale objects attached to two semi-infinite current/voltage electrodes, thereby solving the scattering potential at finite bias. The central quantity of SMEAGOL is the retarded Green function of the molecule– electrode system, which is defined by direct inversion of the following equation:
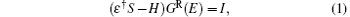
where H is the Hamiltonian matrix, S is the corresponding overlap matrix, I is the infinitely-dimensional identity matrix, ε † = limδ → 0 + E + iδ , and E is the energy. During the calculations, the electronic structures are assumed to be expandable over a local orbital basis set, similar to SIESTA and SMEAGOL. The efficient electron screening in the metallic electrodes causes their effects on the scattering region to be incorporated into the self-energies iR [i = left (L) or right (R)]; hence, the retarded Green function of the scattering region can be calculated as follows:
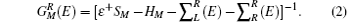
Using the standard NEGF procedure, [29– 31] the non-equilibrium charge density and a new Kohn– Sham Hamiltonian can be computed from the retarded Green’ s function. Hence, a self-consistent procedure is set to evaluate the scattering potential in the presence of an external bias. As the self-consistent cycle converges, the conductance, G, is obtained from the Landauer formula given by
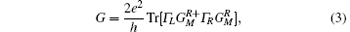
where the Γ ’ s are the anti-Hermitian parts of the self-energies, e is the electron charge, and h is the Planck constant. Finally, the two-terminal current I can be calculated by integrating the conductance over the bias window [Eq. (4)]:
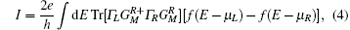
where f is the Fermi function and μ L/R is the chemical potential for the left/right electrode. In-depth calculation details can be found in the original references.[30, 31]
DFT calculations utilize the Perdew– Zunger form[36] of the local-density approximation (LDA) to the exchange– correlation functional and a double-zeta plus polarization basis set for C, S, and Au. Scalar-relativistic Troullier– Martins pseudopotentials[37] in non-local form are generated from the 5d106s1 reference configuration for Au, 3s23p4 configuration for S, and 2s22p2 configuration for C. Periodic boundary conditions are used in the basal plane (i.e., orthogonal to the transport direction) with four irreducible k-points in the two-dimensonal (2D) Brillouin zone. The z-component of k is important for one-dimensional (1D) systems, in which the z-direction corresponds to the transport direction. The conductance is defined as the total transmission at the Fermi energy. In the calculations, the conductance is computed by the summation of all transmissions over all k and Fermi energies, and the average is taken over all transmissions at all possible k and Fermi energies. A k-grid sampling (dimensions, 2 × 2 × 100; mesh cut-off, 200 Ry, 1 Ry = 13.605 eV) for the gold electrodes is employed. The same mesh cut-off is also used in self-consistent transport calculations. A total of 128 real and 50 complex energy points are considered during integration of the Green function.
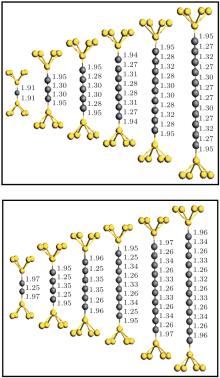
We have calculated the combined electrode– chain system containing the chain itself together with seven and six Au (100) atomic layers, each containing 3 × 3 atoms in the left- and right-hand surface planes. Periodic boundary conditions are imposed on all directions. The system is divided into three regions to consider the molecule– electrode coupling and electrode screening effect: the left electrode, the right electrode, and the scattering region that also includes portions of the two other electrodes. The optimal configuration of the chains is determined via the conjugated gradient method, as well as by relaxing chain coordinates and the positions of the 5Au atoms (pyramid) binding the chain in each lead (Fig. 1). Results of geometry optimization for chains of different lengths are shown in Fig. 1. A locally stable configuration is found by changing the distance between the end atoms of the chain and apices of the point contacts.
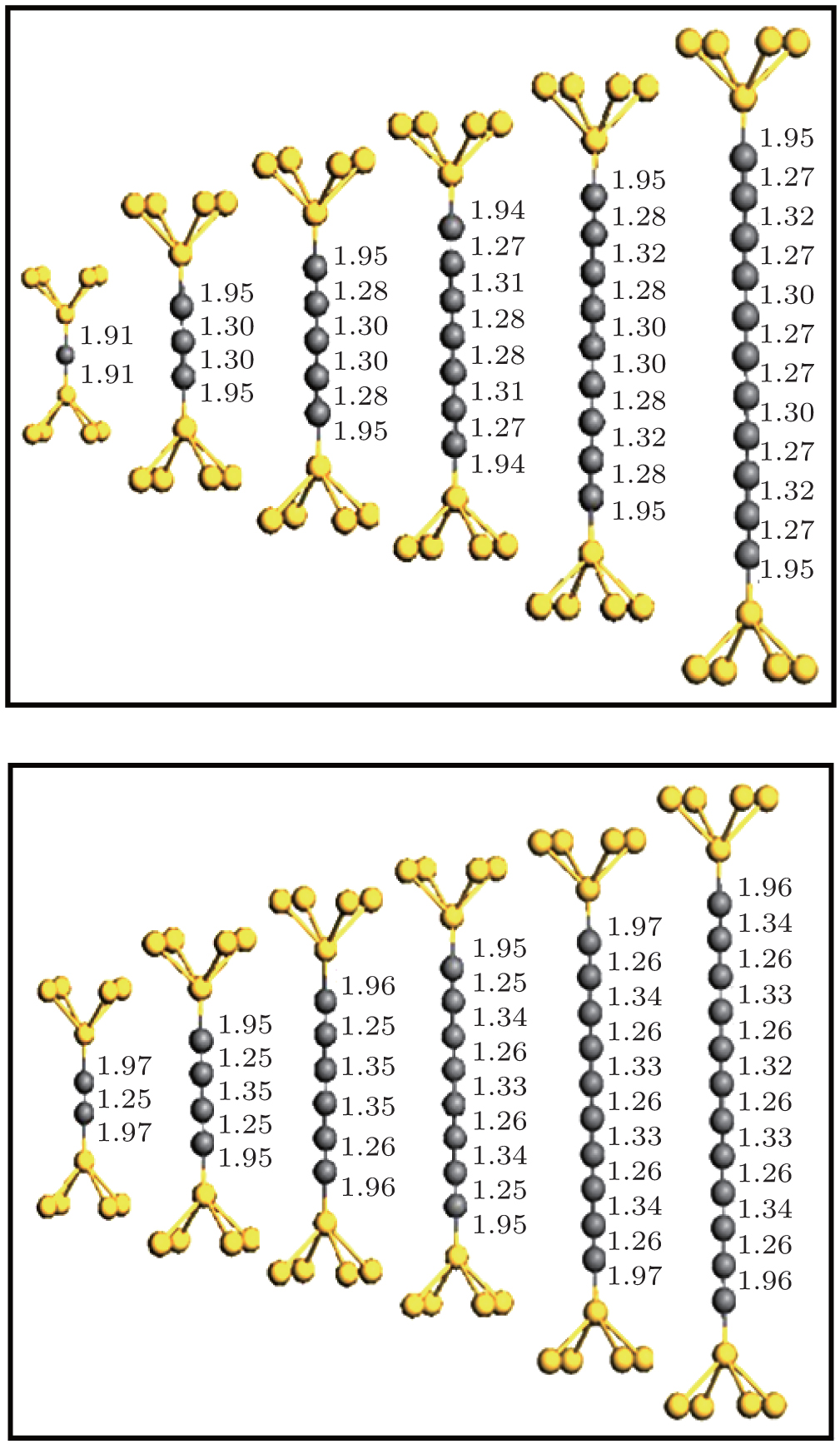 | Fig. 1. Structure of a molecular device in our simulations (only the carbon chain capped with the five pyramid Au atoms). Five atoms on each side represent Au atom, the middle atoms refer to C atoms. The upper panel corresponds to odd carbon atomic chains and the lower panel corresponds to even carbon atomic chains. |
In general, all structures maintained mirror symmetries after relaxation with respect to a plane passing through the center of the chain parallel to the electrode surface. The C– C bond lengths in the chain strongly depend on the even or odd parity of the chain; this condition is similar to that for the structure of monoatomic carbon chains covalently connected to grapheme nanoribbons.[13] On the one hand, even chains exhibit dimerization, particularly an alternation between short bond (1.25± 0.01 Å ) and long bond (1.34± 0.01 Å ), and can thus be considered as a polyyne-like structure. On the other hand, the bonds of length 1.29± 0.02 Å are equal in the middle of odd chains. These chains have smaller bond length alternations near end atoms and thus acquire a cumulene-like property. The Au– C distance (1.95± 0.02 Å ) between the apex atom and chain remains larger than the interatomic distances in the chain. This condition indicates that bonding within the chain is stronger than bonding to the electrodes.
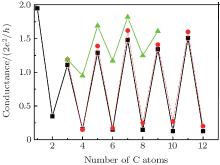
We then investigate electronic transport through CACs using previously optimized atomic conformations and bias voltages, Vbias, at 0 V. The behavior of the even– odd conductance oscillation in CACs is shown in Fig. 2. One of the most striking features of monatomic wires is the nonmonotonic behavior of conductance as a function of the number of atoms in the wire.[38, 39] Such a behavior has been predicted by Lang[39] for carbon atomic wires linking a pair of metal electrodes. The values of conductance G of atomic wires fall in an approximate range of G0 to 2G0 for ballistic transport. A regular even– odd conductance oscillation is also observed in our calculation for CACs linking Au (100) electrodes, but the conductance values are different. Odd-numbered CACs have a conductance near 1.2 G0 (quantum unit G0 = e2/π h), whereas even-numbered chains have a lower conductance. This feature is illustrated in Fig. 2 (black square).
 | Fig. 2. Conductances as a function of the number of atoms in the carbon atomic capped with the ends carbon atoms (square) and sulfur atoms (circle), respectively. Triangle represents the results of Lang. |
The conductance of CACs capped with sulfur atom linking Au (100) electrodes is determined to compare conductance stabilities. Similar conductance oscillation characteristics are observed, and the amplitude of the conductance oscillation is delicately influenced by several factors. This feature, which is relatively stable based on a thiol joint between the chain and leads, is illustrated in Fig. 2 (circle). The following study focuses on the electron transport properties of carbon chains directly connected between Au (100) electrodes with sp3 bonding at the molecular junction joint. A large and complex couple exists between single C atoms and conducting electrons if the carbon chain contains only one or two C atoms. Therefore, to investigate the oscillation characteristics of CACS, the smallest size used is a C3 chain.
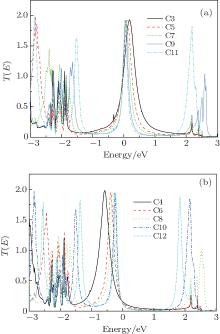
The transport mechanism can be further explained in terms of the transmission spectrum. Figure 3 shows the transmission spectra T(E; V) of all the models under zero voltage. A common characteristic of odd- and even-numbered chains is that more transmission peaks appear in the transport spectra as the CACs become longer. This case is due to the interaction of more atoms in the molecular device.[40] A clear difference also exists between odd- and even-numbered chains. A significant shift in the highest occupied molecular orbital (HOMO) resonance toward higher energies is observed as the number of carbon atoms increases in even-numbered chains. This shift moves the HOMO closer to the EF. However, as the EF is still located at the tail of the transmission peak, the current (discussed below) and conductance remain relatively small. The Fermi energy in odd-numbered chains is close to the lowest unoccupied molecular orbital, which is the main transmission channel. However, the HOMO is an important transmission channel with a broad transmission peak in chains C9 and C11.
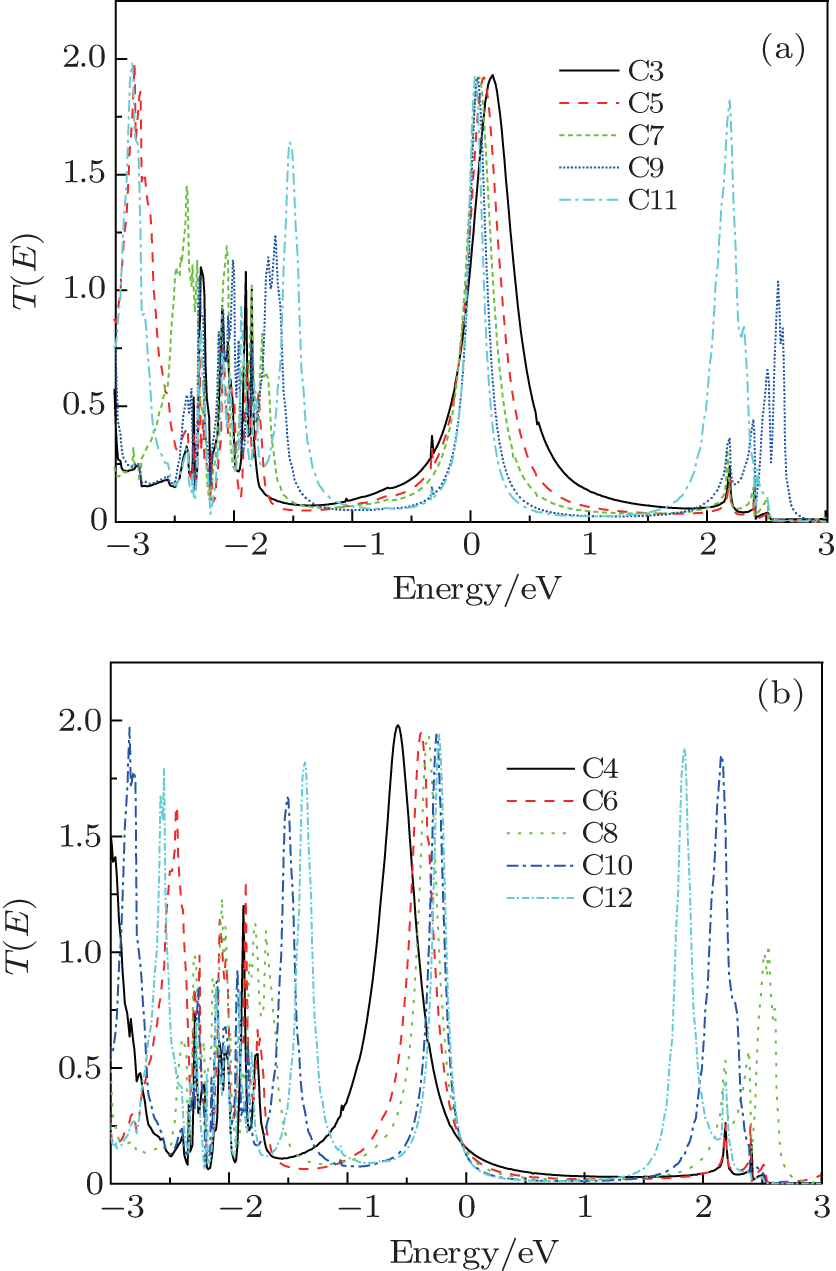 | Fig. 3. Transmission spectra of the odd-numbered carbon (a) and the even-numbered carbon (b) at zero voltage. |
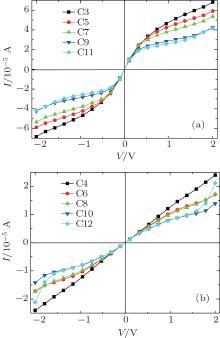
A system is driven out of equilibrium when the bias voltage is applied to it. The I– V characteristics of all models at bias voltages ranging from − 2.0 V to 2.0 V are shown in Fig. 4. Several important features in the odd and even chains are clearly visible: (i) odd chains have almost identical I– V characteristics over the whole bias voltage range, and currents dramatically increase when the voltage is less than 0.2 V. The currents continuously increase as the bias is increased further but the rate of this increase is fairly low. (ii) Unlike the case in odd chains, the currents of even chains increase slowly over a wide range of bias voltages but longer CACs show larger current increases near 2 V.
 | Fig. 4. Variations of current I with applied bias V for odd carbon atomic chains (a) and even carbon atomic chains (b), respectively. |
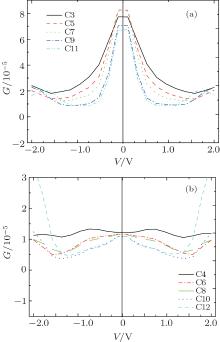
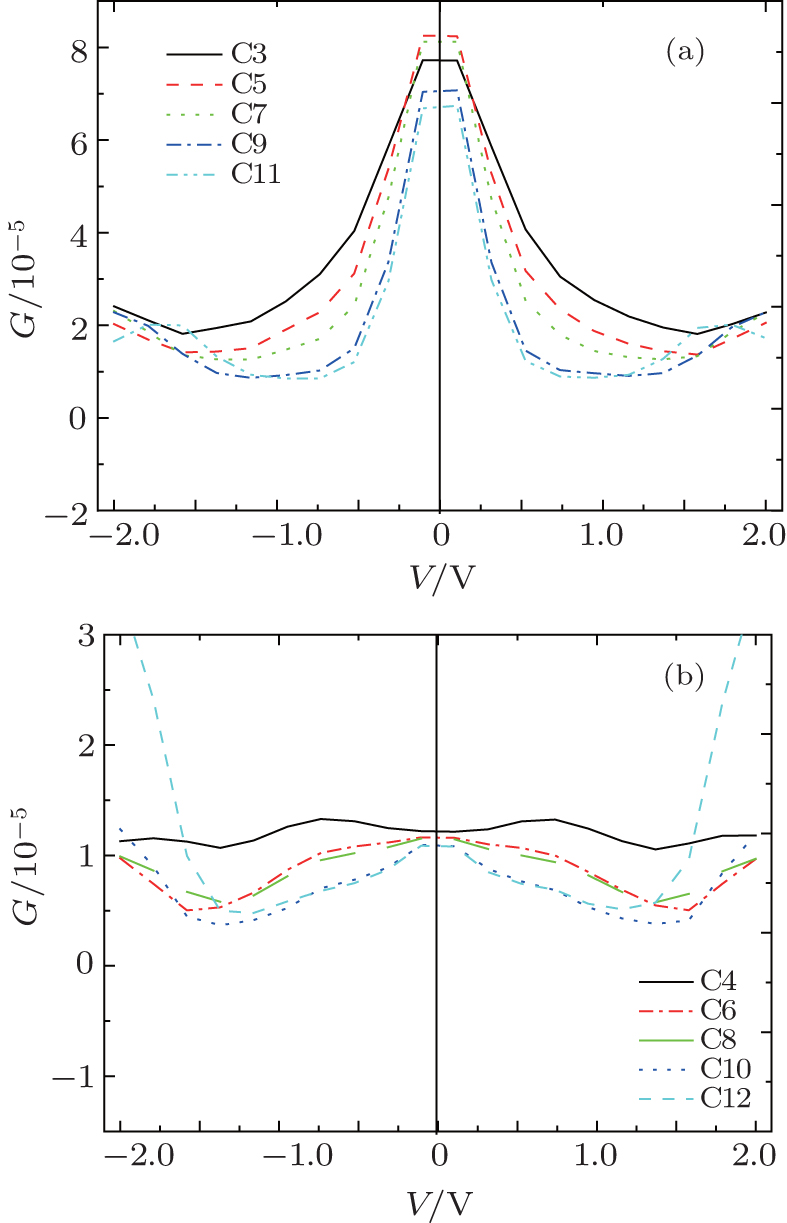 | Fig. 5. The second derivative of the conductance d2G(0)/d2V as a function of the number of atoms. Variations of differential conductance G(V) = dI/dV with applied bias V for odd carbon atomic chains (a) and even carbon atomic chains (b), respectively. |
We calculate the differential conductance using the I– V curve (Fig. 5). For relatively short even-numbered chains, such as those of C4, C6, and C8, the conductance varies by less than 5% for bias voltage up to ∼ ± 0.75 V, which can be considered as a linear response regime. The conductance decreases monotonically for bias voltage up to ∼ 1.5 V. However, no linear response regime exists for chains of C10 and C12, and the conductance decreases monotonically from 0 V to 1.5 V. The conductance for odd-numbered chains is nearly invariant for bias voltage up to ∼ ± 0.2 V and decreases monotonically for bias voltage up to ∼ 1.5 V.
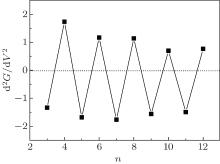
An important characteristic of the conductance curve that can be measured experimentally is its second derivative at the Fermi energy. If the transmission near the Fermi energy is approximated by the function T(E) = T(0) + T(0)″ E2, the differential conductance is G(V) = G0[T(0) + T(0)″ V2/4]. The second derivative of the conductance can be given by d2G(0)/d2V = T(0)″ /2[41] because odd– even conductance oscillation of the CACS is the resonance behavior of the peaks during transmission. Thus, the second derivative of the conductance must be positive for even-numbered chains and negative for odd-numbered wires at the Fermi energy. The relevant results are shown in Fig. 6.
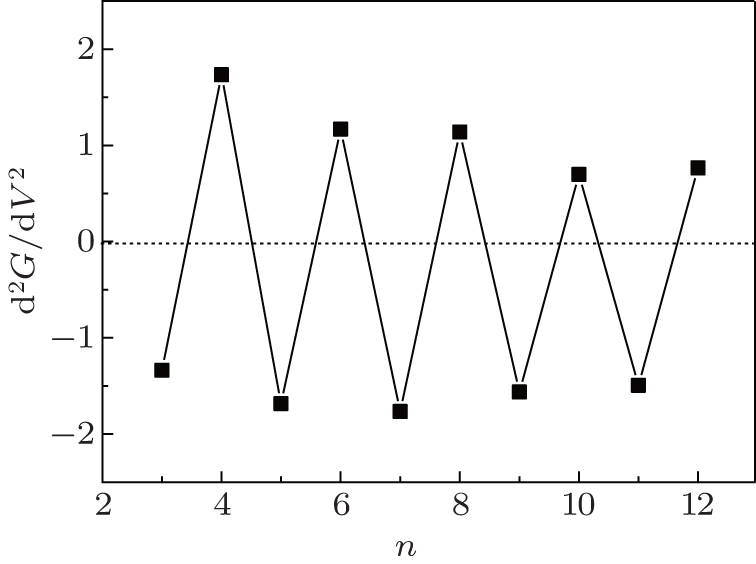 | Fig. 6. The second derivative of the conductance d2G(0)/d2V as a function of the number of atoms. |
We examine the electronic transport properties of CACs attached to Au (100) electrodes using density functional theory and the non-equilibrium Green’ s function method. We also simulate the behavior of even– odd conductance oscillation in CACS. Even– odd conductance oscillation is delicately influenced by changes in the end carbon or sulfur atoms and variations in coupling strength between the chain and leads. The amplitude of the conductance oscillation is predicted to be relatively stable based on a thiol joint between the chain and leads. We also calculate I– V characteristics over bias voltages ranging from − 2.0 V to 2.0 V and determine the differential conductance and second derivative of the conductance using the I– V curves obtained. The calculated results show clear differences between odd-numbered and even-numbered CACs.
All of the authors thank Dr. Sanvito for providing us with the SMEAGOL code.
| 1 |
|
| 2 |
|
| 3 |
|
| 4 |
|
| 5 |
|
| 6 |
|
| 7 |
|
| 8 |
|
| 9 |
|
| 10 |
|
| 11 |
|
| 12 |
|
| 13 |
|
| 14 |
|
| 15 |
|
| 16 |
|
| 17 |
|
| 18 |
|
| 19 |
|
| 20 |
|
| 21 |
|
| 22 |
|
| 23 |
|
| 24 |
|
| 25 |
|
| 26 |
|
| 27 |
|
| 28 |
|
| 29 |
|
| 30 |
|
| 31 |
|
| 32 |
|
| 33 |
|
| 34 |
|
| 35 |
|
| 36 |
|
| 37 |
|
| 38 |
|
| 39 |
|
| 40 |
|
| 41 |
|