{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Structural, electronic, and magnetic properties in FeAlAu𝓷 (𝓷 = 1–6) clusters: A first-principles study*
[Zhang Jian-Feia) , Zhang Menga)†  , Zhao Yan-Wei
, Zhao Yan-Weia) , Zhang Hong-Yua) , Zhao Li-Nab) , Luo You-Huaa)‡ ]
, Zhao Yan-Wei]
|
|


†Corresponding author. E-mail: mzhang@ecust.edu.cn
‡Corresponding author. E-mail: yhluo@ecust.edu.cn
*Project supported by the National Natural Science Foundation of China (Grant Nos. 11204079, 11304096, and 11404333) and the Natural Science Foundation of Shanghai, China (Grant No. 12ZR1407000).
The geometries, electronic and magnetic properties of the trimetallic clusters FeAlAu n ( n= 1–6) are systematically investigated using density functional theory (DFT). A number of new isomers are obtained to probe the structural evolutions. All doped clusters show larger relative binding energies than pure Au n+2 partners, indicating that doping with Fe and Al atoms can stabilize the Au n clusters. The highest occupied molecular orbital–lowest unoccupied molecular orbital (HOMO–LUMO) gaps, vertical ionization potentials and vertical electron affinities are also studied and compared with those of pure gold clusters. Magnetism calculations demonstrate that the magnetic moments of FeAlAu n clusters each show a pronounced odd–even oscillation with the number of Au atoms.
Gold clusters have been intensively studied during the past two decades due to the potential value of their catalytic, electronic, and optical properties. Pure Aun clusters in the small-to-medium size range have been intensively investigated experimentally and theoretically.[1– 19] In addition, a considerable amount of research work has been carried out on bimetallic gold clusters to enhance the stabilities of gold clusters and extend the potential applications. Most of these works focused on the doping transition-metal (TM) atoms into gold clusters.[20– 29] Some studies on gold clusters doped with non-transition elements have also been carried out.[30– 37] The above work shows that gold clusters doped with a proper element have distinctive structures and interesting properties.
However, studies on both structures and properties of trimetallic nanoclusters are extremely challenging because of the drastically increased complexity due to the presence of a third-part atom. There are numerous changes of structures in FeAlAun clusters. To the best of our knowledge, no systematical theoretical study of MN-doped gold clusters FeAlAun (M = Fe, N = Al) has been reported. In our previous work, [38– 41] the geometries, electronic and magnetic properties of FeAu6 and AlAun clusters were studied using the relativistic all-electron density functional theory. In this work, we extend our investigation on FeAlAun (n = 1– 6) clusters to explore their geometrical structures, electronic and magnetic properties. It is hoped that the present research will be conducible to experimental realizations and verifications on the ternary metal clusters.
Structure optimizations and property calculation in this work are performed using DFT implemented in the DMol3 package.[42] Relativistic calculations are carried out via pseudopotentials with scalar relativity (VPSR) to valence orbitals relevant to atomic bonding properties. All-electron spin-unrestricted calculations with double numerical plus d-functions (DND) are employed. Generalized gradient approximation in the Perdue– Burke– Ernzerhof (PBE) functional form is chosen.[43] Spin multiplicity ranging from 1 to 8 of this system is considered. The convergence threshold for the maximum energy change is set to be 2× 10− 5 Hartree (1 Hartree = 27.2114 eV) in the geometry optimization. The convergence thresholds are set to be 0.004 Hartree/Å for the force, and 0.005 Å for the displacement. Harmonic vibrational frequency analysis is also carried out at the same level of theory to confirm that the energies of isomers are truely minimal values.
The quality of the current computational method (PBE/DND/VPSR) is tested by the calculations on Au2, 
Table 1. Calculated ground-state parameters and properties of Au2,  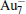 |
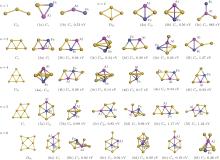
To discover the stable structures of FeAlAun (n = 1– 6) clusters, we use the computational methods described in Section 2 and consider extensive two-dimensional (2D) and three-dimensional (3D) structures to optimize each FeAlAun cluster structure. The obtained lowest-energy structures and some low-lying isomers are shown in Fig. 1. The symmetries and the differences in total energy between the lowest-energy and the low-lying isomers are listed. The lowest-energy structures of the corresponding bare Aun+ 2 clusters are also given in Fig. 1 for comparison. The optimized geometries reveal that small pure gold clusters prefer 2D structures, and the ground-state structures of FeAlAun clusters are all 2D configurations, except for FeAlAu4. A single Au atom edge-capped structure of the FeAlAun− 1 cluster is a dominant growth pattern. Previous studies showed that unlike alkali metal clusters, pure Aun clusters favor 2D structures due to the strong relativistic effect of Au, and adding one Au atom to the Aun− 1 cluster is its main structural evolution for its small size.[7, 44] The tendency to retain the overall structural integrity of the gold clusters is the possible explanation of the growth pattern in FeAlAun.
 | Fig. 1. Lowest-energy structures and low-lying isomers with the relative energies (in unit eV) of FeAlAun (n = 1– 6) clusters, optimized with PBE/DND/VPSR (DMol3). The ground-state geometries of the corresponding bare Aun + 2 clusters are also given on the left. See Table 2 for the corresponding energetic and structural information. Red, gray, and yellow circles represent aluminum, iron, and gold atoms, respectively. |
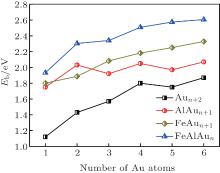
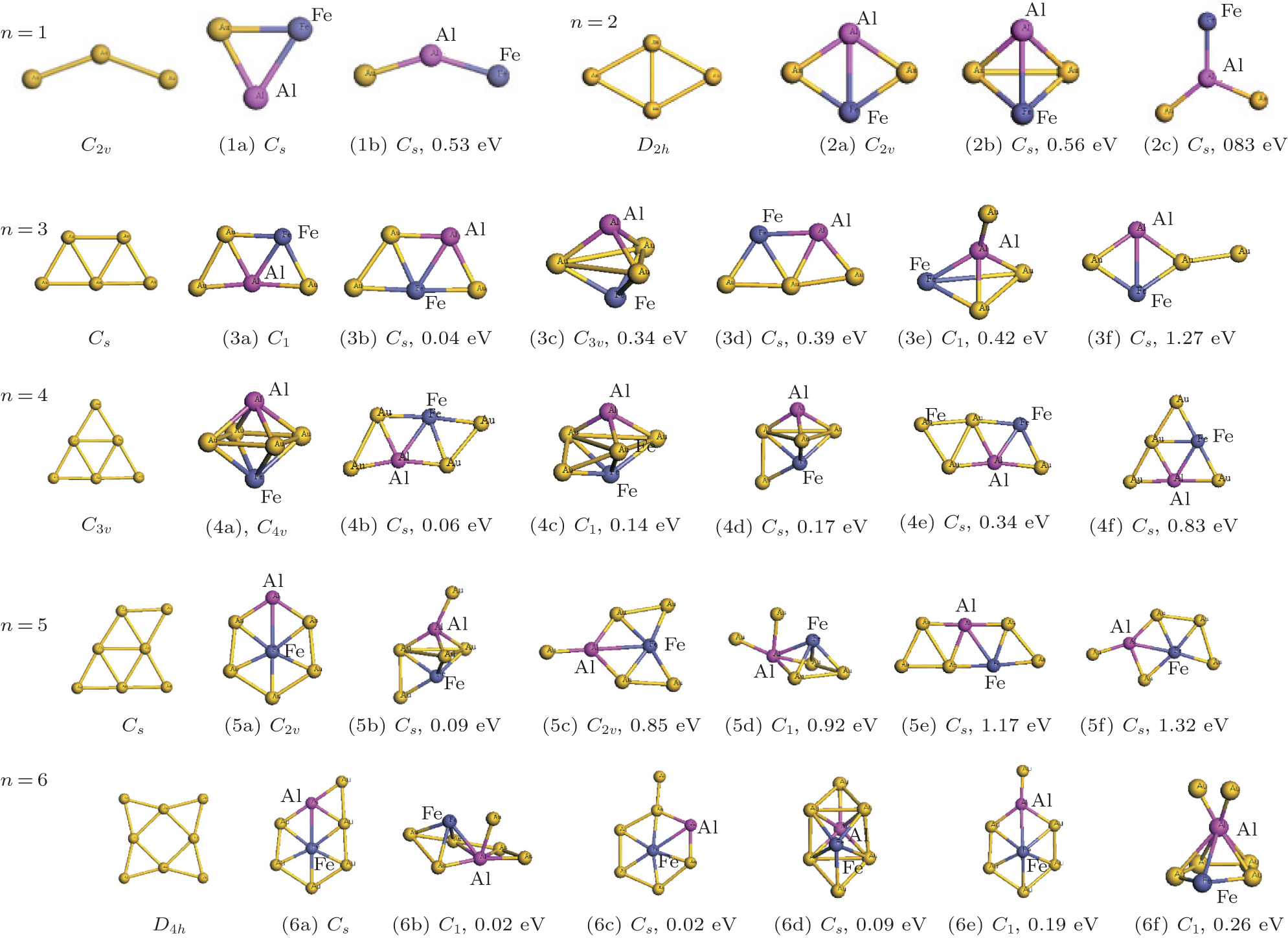
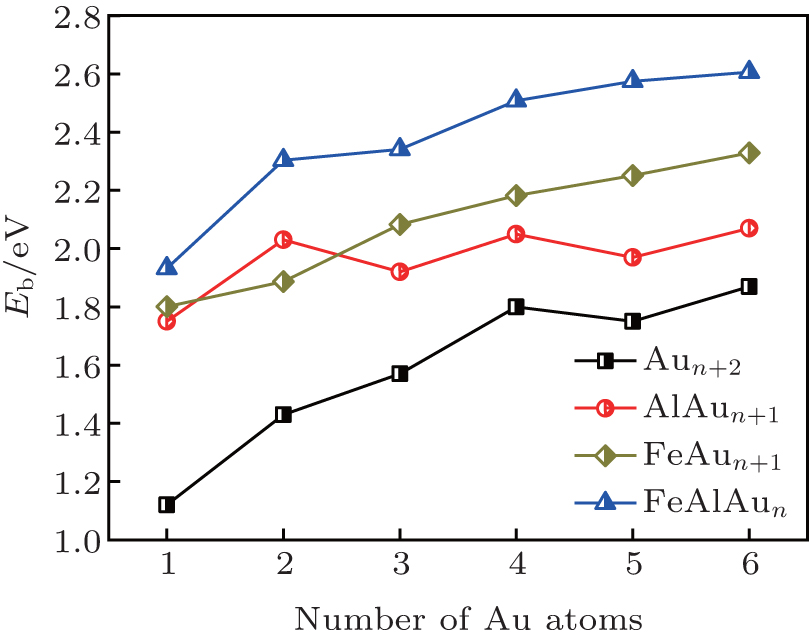 | Fig. 2. Average binding energies per atom (Eb) of the FeAlAun, AlAun + 1, FeAun + 1, and bare Aun+ 2 clusters (n = 1– 6) for the lowest-energy structures. |
| Table 2. Values of HOMO– LUMO energy gap Egap (eV), vertical ionization potential VIP (eV), vertical electron affinity VEA (eV), and atomic charges (in atomic unit a.u.) on Fe and Al atoms of FeAlAun (n = 1– 6) clusters, for the lowest-energy structures. |
The triangle structure (1a) with Cs symmetry is found to be the most stable structure for the FeAlAu cluster. The angular isomer (1b) with C2v symmetry is 0.53 eV higher in energy than the lowest-energy structure. The most stable structure of the FeAlAu2 cluster is a C2v rhombic planar structure (2a), which relates to the lowest-energy structure of the Au4 cluster. The 3D geometries are all higher in energy. Like the lowest-energy structure of a pure Au5 cluster, the most stable structure of FeAlAu3 is a planar trapezoid configuration (3a), which can be obtained by adding one Au atom to the structure (2a). Another isomer (3b) with an Fe atom sitting in the center is only 0.04 eV higher in energy than the structure (3a). A 3D trigonal bipyramid (3c) with C3v symmetry is 0.34 eV higher in energy than the structure (3a). Unlike other mall size of FeAlAun (n = 1– 6) clusters, the ground-state structure (4a) of the FeAlAu4 cluster is not a planar structure, but a 3D quadrilateral bipyramid structure with C4v symmetry. The Fe and Al impurities occupy the two opposite apex positions. The first low-lying isomer (4b), which is a planar geometry with Cs symmetry by subsequent addition of one Au atom to isomer (3a), is only 0.06 eV higher than the most stable structure. The lowest-energy structure of the FeAlAu5 cluster (5a) is a planar irregular hexagon with C2v symmetry. The Al atom occupies the top position, and the Fe atom occupies the central position. Figure 1 shows that the 3d Fe atom in FeAlAu5 and FeAlAu6 is likely to be surrounded by the host gold atoms. The hexagon structure can be seen as a distorted isomer of the ground-state structure of FeAu6 with an Fe atom sitting in the center of the Au6 ring.[40] To further analyze the stabilities of clusters, the average binding energies per atom (Eb) of the FeAlAun, AlAun+ 1, FeAun+ 1, and bare Aun+ 2 for the lowest-energy structures are calculated and depicted in Fig. 2 each as a function of the number of Au atoms. It can be seen from Fig. 2 that the values of Eb for the ground state of FeAlAun clusters are significantly higher than those of the corresponding pure Aun+ 2, AlAun+ 1, and FeAun+ 1 clusters, respectively. This indicates that doping with two different types of metal atoms (Al and Fe) can effectively enhance the stabilities of the Aun clusters.
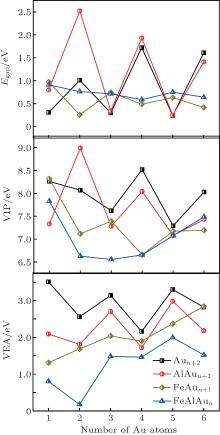
 | Fig. 3. Variations of HOMO– LUMO energy gap Egap (eV), vertical ionization potential VIP (eV), and vertical electron affinity VEA (eV) with the number of Au atoms, of the FeAlAun, AlAun+ 1, FeAun+ 1, and bare Aun+ 2 clusters (n = 1– 6) for the lowest-energy structures. |
According to the frontier molecular orbital theory, to a certain extent, the energy gap between HOMO and LUMO determines the charge transfer in a cluster, which affects the chemical activity. The vertical ionization potential (VIP) and the vertical electron affinity (VEA) reflect the capacities of losing or gaining electrons, respectively. The calculated results of Egap, VIP, VEA, and the atomic charge on Fe and Al atoms of the lowest-energy structures of FeAlAun (n = 1– 6) clusters are given in Table 2 and plotted in Fig. 3. These electronic properties of the lowest-energy structures of AlAun+ 1, FeAun+ 1, and pure Aun+ 2 clusters are also plotted in Fig. 3 for comparison. It can be seen from Fig. 3 that there are obvious odd– even oscillations in the spectra of the Egap, VIP, and VEA in AlAun+ 1, FeAun+ 1, and pure Aun+ 2 clusters, while this phenomenon disappears in FeAlAun clusters. Both VIPs and VEAs of FeAlAun clusters are smaller than those of the AlAun+ 1, FeAun+ 1, and pure Aun+ 2 clusters. It reveals that the electronic structures of FeAlAun clusters are more unstable, and doping Fe and Al atoms into Aun clusters can enhance the chemical activities of the gold clusters.
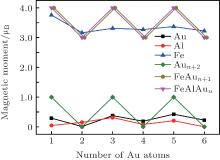
In what follows, we will discuss the magnetic properties of the FeAlAun clusters. The local magnetic moments of Fe, Al, and Au atoms, and total magnetic moments of FeAlAun clusters for the lowest-energy structure are listed in Table 3 and plotted in Fig. 4. The Mulliken population analysis in Table 3 shows that the total magnetic moment of the clusters is localized mainly in the Fe atom. A small quantity of magnetic moment is found in the Al atom and host Au atoms, which shows that the 3d Fe atom plays a decisive role in the magnetic moment of Au clusters. The total magnetic moment of bare Aun+ 2 clusters shows a pronounced odd– even alternation (0 μ B and 1 μ B, respectively) with the number of Au atoms. A single Fe atom has four unpaired electrons with an atomic configuration of 3d64s2, and the d electrons are localized. The electronic shell structure of a single Au atom is 5d106s1. When an Fe atom with the partially filled 3d orbital is doped into Aun clusters, one unpaired s electron from the Aun clusters will reduce an offset of 1 μ B magnetic moment of the Fe atom. So the formed FeAlAun clusters generate a large magnetic moment (3 μ B or 4 μ B) with the even or odd number of Au atoms. This suggests that the FeAlAun clusters have potential applications in new nanomaterials with tunable magnetic properties by adding even or odd number of Au atoms.
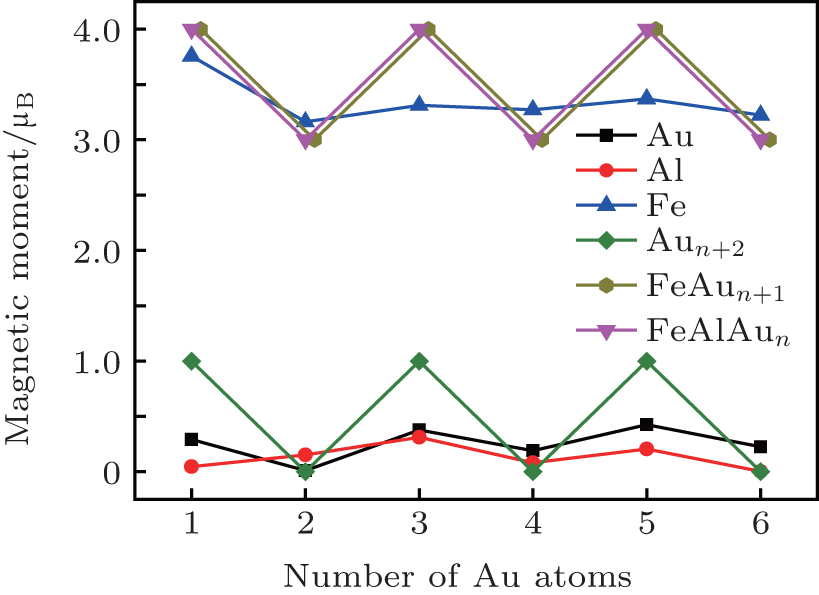 | Fig. 4. Local magnetic moments of Fe, Al, Au atoms, and total magnetic moments of FeAlAun, FeAln+ 1, and bare Aun+ 2 (n = 1– 6) clusters for the lowest-energy structures. |
| Table 3. Local magnetic moments of Fe, Al, and Au atoms, and the total magnetic moment of FeAlAun (n = 1– 6) clusters for the lowest-energy structures. |
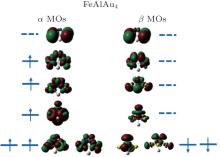
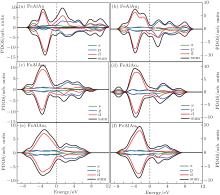
In order to explain the phenomenon of the magnetic properties in FeAlAun, figure 5 shows an example of the schematic molecular orbitals (MOs) and MO energy level correlation diagram of the FeAlAu4. The d orbit characteristic of an Fe atom is marked in the figure. Two d orbitals (dxz and dyz) of the Fe atom are degenerate and occupied both by α electrons (spin-up) and β electrons (spin-down), and other three orbitals dz2, dxy, and dx2 − y2, are singly occupied by three α electrons with spin-up, respectively, resulting in 3 μ B of the total magnetic moment in FeAlAu4. In addition, we also give the partial densities of states (PDOSs) in Fig. 6. It can be found that the shapes of α and β PDOSs of s, p, and d orbitals for FeAlAun clusters are always different, which explains why these FeAlAun each have a large magnetic moment. Figure 6 also shows that the electronic states below the Fermi level are contributed mainly from the d state.
 | Fig. 5. Molecular orbitals (MOs) and their energy level correlation diagram of the FeAlAu4 for the lowest-energy structures. Continuous lines correspond to the filled levels and the dotted lines correspond to the unfilled states. The surface isovalue for molecular orbital plotting is 0.02 e/Å 3. |
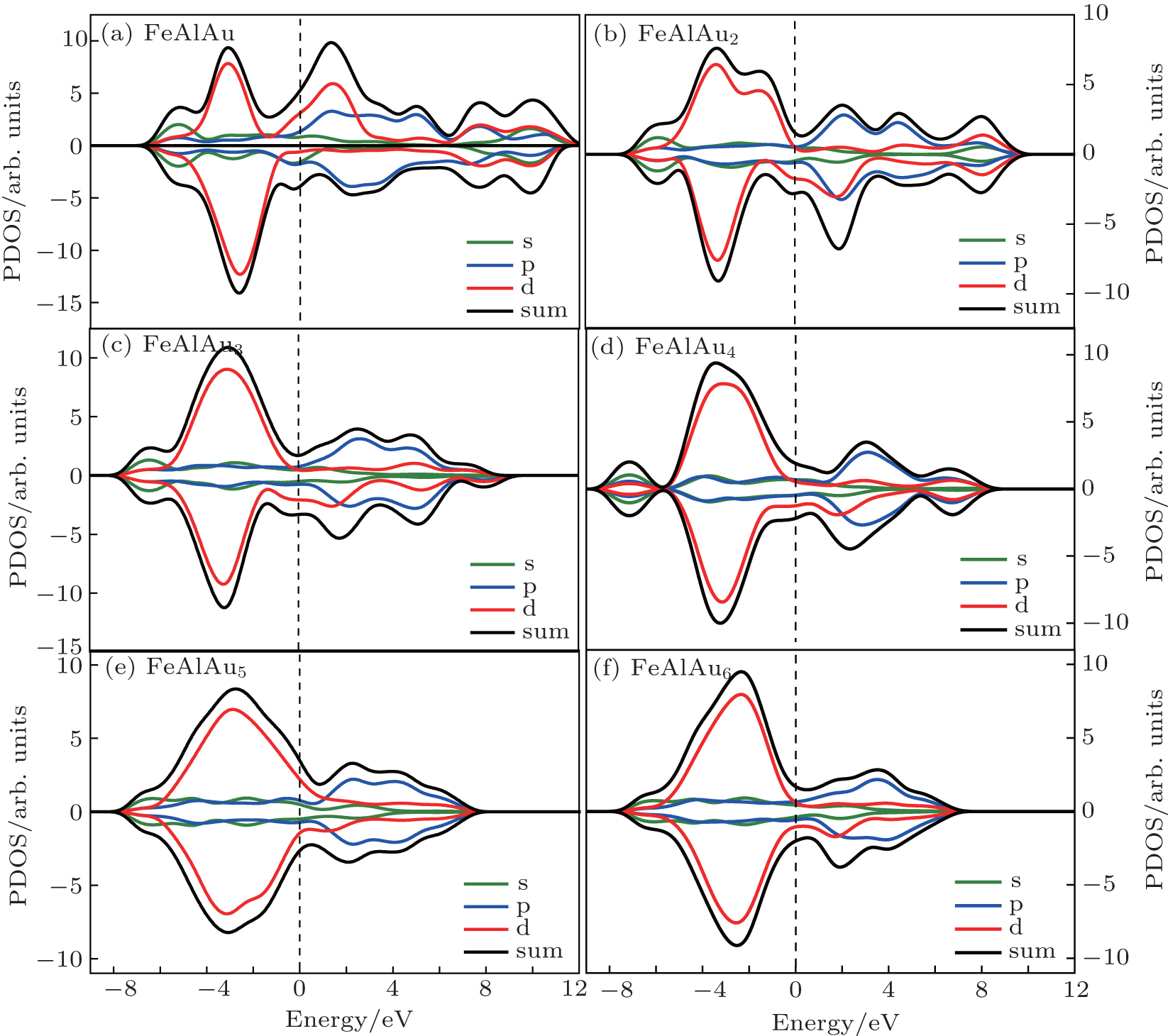 | Fig. 6. PDOSs of s, p, and d orbitals for FeAlAun (n = 1– 6) clusters. Spin-up (positive) and spin-down (negative) densities are given in each case. The dashed lines indicate the location of the Fermi level. |
A density functional theory is investigated to study the geometric structures, relative stabilities, electronic and magnetic properties of the ternary metal clusters FeAlAun (n = 1– 6). A number of new isomers are obtained and discussed. All the lowest-energy structures of FeAlAun clusters prefer planar structures, except for the FeAlAu4 cluster. It is found that a single Au atom edge-capped structure of the FeAlAun− 1 cluster is a dominant growth pattern. Doping with Al and Fe atoms can greatly enhance stabilities of geometric structures, but reduce stabilities of electronic structures in gold clusters. FeAlAun clusters have tunable magnetic properties of odd– even oscillations by adding an even or odd number of Au atoms. This suggests that the stable FeAlAun clusters may have the potential to be utilized in new nanomaterials as building blocks.
| 1 |
|
| 2 |
|
| 3 |
|
| 4 |
|
| 5 |
|
| 6 |
|
| 7 |
|
| 8 |
|
| 9 |
|
| 10 |
|
| 11 |
|
| 12 |
|
| 13 |
|
| 14 |
|
| 15 |
|
| 16 |
|
| 17 |
|
| 18 |
|
| 19 |
|
| 20 |
|
| 21 |
|
| 22 |
|
| 23 |
|
| 24 |
|
| 25 |
|
| 26 |
|
| 27 |
|
| 28 |
|
| 29 |
|
| 30 |
|
| 31 |
|
| 32 |
|
| 33 |
|
| 34 |
|
| 35 |
|
| 36 |
|
| 37 |
|
| 38 |
|
| 39 |
|
| 40 |
|
| 41 |
|
| 42 |
|
| 43 |
|
| 44 |
|