

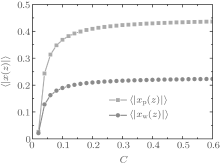
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A theoretical investigation on anomalous switching of single-stranded deoxyribonucleic acid (ssDNA) monolayers by water vapor*
[Zhao Xin-Juna)†  , Gao Zhi-Fu
, Gao Zhi-Fub) , Jiang Zhong-Yinga) ]
, Gao Zhi-Fu|
|


†Corresponding author. E-mail: zhaoxinjunzxj@163.com
*Project supported by the National Natural Science Foundation of China (Grant Nos. 21264016, 11464047, and 21364016), the National Basic Research Program of China (Grant No. 2012CB821500), and the Natural Science Foundation of Xinjiang Uygur Autonomous Region, China (Grant No. 2013211A053).
In this paper, we use a molecular theory to study the anomalous switching of ssDNA monolayers. Here, both ssDNA–water and water–water hydrogen bonds and their explicit coupling to the ssDNA conformations are considered. We find that hydrogen bonding becomes a key element in inducing the anomalous switching of ssDNA monolayers. This finding accords well with the experimental observations. Based on our theoretical model, we predict that the anomalous switching induced by water vapor will be applicable to a wide range of hydrogen bonds polymers, and ssDNA–water hydrogen bonds and water–water hydrogen bonds hybridization will lead to the hydrogen-bond network formation of 3D ssDNA monolayers.
In recent years, biosensors composed of monolayers of irreversibly grafted biopolymers (DNA or protein molecules) have attracted considerable attention, both experimentally[1– 5] and theoretically.[6, 7] In particular, DNA monolayers have also been successfully used in the areas of molecular recognition, molecular electronics, [8] and molecular sensing.[9, 10] Such monolayers can be formed by grafting single-stranded deoxyribonucleic acid (ssDNA) or double-stranded DNA (dsDNA) onto a flat gold surface[11– 14] or silicon surface.[15– 17]
It is well established that DNA molecules are negatively charged in solution under physiological conditions (pH ≈ 7). The state of DNA monolayers is determined by the compositions of water, salt, and buffer solution.[18] For instance, cations may induce a deformation of DNA self-assembled monolayers, [19, 20] and pH can induce a transition from an expanded state to a collapsed state of ssDNA monolayers.[7, 21] However, an experiment, performed by Mertens et al., [22] demonstrated that the states of single-stranded DNA (ssDNA) and single-stranded (ssPNA) monolayers are directly controlled by changing the concentration of water– vapor. According to experimental results of Ref. [22], anomalous changes in the monolayer tension were detected: the monolayers would first shrink and then swell as a result of hydration, which implied an anomalous switching of ssDNA and ssPNA monolayers. In addition, Mertens et al.[22] also observed anomalous switching in neutral ssPNA monolayers, which suggested that the electrostatic interactions are not dominated. On the other hand, the results revealed that there exists a hysteresis in the swelling of collapsed ssDNA and ssPNA chains. Motivated by these intriguing results, Wagman et al.[23] made a careful analysis of the response of a grafted ssDNA monolayer to variation of water– vapor concentration using a mean field theory. According to the model of Ref. [23], the switching anomaly was only considered as a direct result of monomer– monomer repulsion and monomer– water attraction. In fact, the DNA hydration is linked to the formation of DNA hydration shells, in which water molecules bind ssDNA chains through hydrogen bonds.[22, 24, 25] Water in DNA hydration shells shows differences between its properties and those of water in bulk solution.[22] Interestingly, Liu and Zhang[26] reported a hysteresis in the cooling process of poly(N-isopropylacrylamide) (PNIPAM) brushes.[26] Cheng et al.[27] attributed the hysteresis to an additional hydrogen bond formed in a collapsed state. According to the researches and analysis above, we think that the conformation of ssDNA chains is significantly influenced by the formation of hydrogen bonds. However, up to now, few theories have been devoted to a molecular-level understanding of switching anomaly and hysteresis caused by the formation of hydrogen bonds in ssDNA monolayers. Thus, it is necessary for us to include more molecular details to properly describe switching anomaly and hysteresis in ssDNA systems though hydrogen bonding.
In this paper, we will use a molecular theory[28– 31] to investigate the switching anomaly of ssDNA monolayers. Here, the formation of hydrogen bonds is introduced following the ideas of Dormidontova[32] and Ren et al.[33] Our aim is to explore how hydrogen bonds influence the conformations of ssDNA monolayers, and to explain the mechanism of switching anomaly and hysteresis of ssDNA monolayers.
The remainder of this paper is organized as follows. In Section 2, we will describe a molecular theoretical model in which both ssDNA– water and water– water hydrogen bonds are taken into consideration. In Section 3, we will present the results and discussion of investigating the effects of both ssDNA– water and water– water hydrogen bonds and their explicit coupling to the ssDNA conformations. Finally, a summary and conclusions are given in Section 4.
We describe a molecular theoretical model that considers the size, shape, and conformation of each ssDNA molecular type with the explicit inclusion of hydrogen bonding. In order to create an ssDNA monolayer system, we assume that ssDNA chains are homogeneously grafted onto the substrate surface defined as the x– y plane at z = 0. The ssDNA monolayer system contains NP ssDNA chains immersed in water– vapor and air, here NP is the number of ssDNA chains. The ssDNA chains are tethered onto a substrate surface, which can be allowed only in the z ≥ 0 half-space. The number of tethered chains per unit area is σ = Np/A. Each ssDNA chain has N segments, and each segment has a volume vp . The numbers of water– vapor and air molecules are Nw and Na, respectively. The volumes of each water– vapor molecule and each air molecule are vw and va, respectively. We also assume that the only the inhomogeneous direction is the direction perpendicular to the substrate surface, that is, the z direction. The free energy per unit area of ssDNA monolayer system of water– vapor and air molecules has the following form:
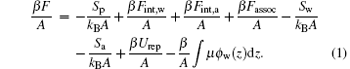
The first term on the right-hand side of Eq. (1) denotes the conformational entropy of ssDNA chains, which can be given by
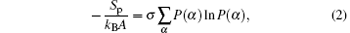
where P(α ) is the probability distribution function of finding a chain in conformation α . Given a probability distribution function, we can calculate any average thermodynamical structural quantity of polymers.[33, 34] The ssDNA volume fraction profile is determined by
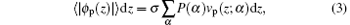
where vp (z; α ) denotes the volume that an ssDNA chain in conformation α contributes in the layer between z and z + dz.
The second and third terms on the right-hand side of Eq. (1) represent the effective intermolecular interaction between ssDNA segments and water– vapor molecules, and the effective intermolecular interaction between ssDNA segments and air molecules, respectively. The final expressions for these two terms are
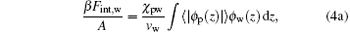 |
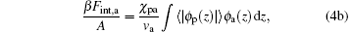 |
where χ pw is the Flory interaction parameter measuring the strength of ssDNA– water effective interaction, and χ pa is the Flory interaction parameter measuring ssDNA-air effective interaction. The ϕ i(z) = ρ i(z)vi (i = w, a) is the position-dependent volume fraction of corresponding free species i, with ρ i(z) denoting the number density of corresponding species i.
The fourth term on the right-hand side of Eq. (1) is the contribution from the formation of hydrogen bonds. According to Refs. [32] and [33], the contribution to free energy arises from the formation of ssDNA– water and water– water hydrogen bonds, and can be written as
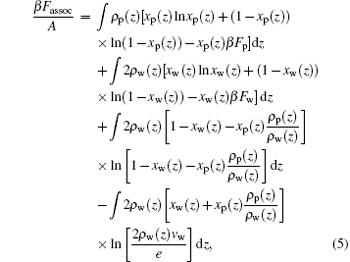
where Fi (i = p, w) is the free energy gain from a single hydrogen bond formation, which includes energetic gain and entropic loss of forming hydrogen bonds.[32] In the equation above, we have introduced two additional variables, the local fraction of ssDNA– water hydrogen bonds, xp(z), and the local fraction of water– water hydrogen bonds, xw(z), which are defined as
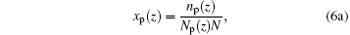 |
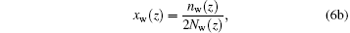 |
respectively, where np(z) and Np(z) are the number of position-dependent ssDNA– water hydrogen bonds and the number of ssDNA chains, respectively; nw(z) and Nw(z) are the number of position-dependent water– water hydrogen bonds and the number of water molecules, respectively. It is also noteworthy that the original definition of Flory interaction parameter χ pw is no longer applicable to the case in which hydrogen bonds are involved. Therefore, it is necessary to introduce an effective interaction parameter χ eff by combining various hydrogen bonds, [32]
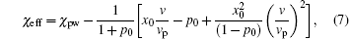
where p0 and x0 describe the associations of ssDNA– water and water– water by hydrogen bonds, respectively. The effective interaction parameter χ eff is of great importance due to its close relation with hydrogen bonds. We also assume that p0 and x0 are proportional to water vapor concentrations: p0 = kpC and x0 = kwC, where kp and kw are corresponding proportional parameters. In order to capture the main qualitative features observed in ssDNA monolayers during hydration, we take the values of kp= 0.75 and kw= 0.001 as reported in Ref. [22].
The effective interaction parameter is related to entropic components and the roles of hydrogen bonds. In this work, we will replace Flory interaction parameter χ pw with the effective interaction parameter χ eff . The temperature dependence of χ pw obeys a standard form of
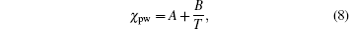
where intrinsically strong hydrogen bonds between complementary base pairs in ssDNA chains are neglected, because we focus on the anomalous switching of ssDNA monolayers by water vapor. The fifth and sixth terms on the right-hand side of Eq. (1) are given by
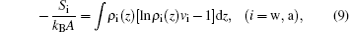
where Sw and Sa denote the z-dependent translational entropies of water vapor molecules and air molecules, respectively. The seventh term on the right-hand side of Eq. (1) represents the repulsive interaction of the system, and can be given by
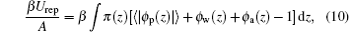
where π (z) is the position-dependent repulsive interaction field, which is determined by packing constraints:
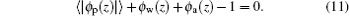
The last term on the right-hand side of Eq. (1) represents the chemical potential of water– vapor molecules, and is determined by
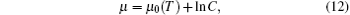
where μ 0(T) is the reference chemical potential depending on temperature T, and C is the bulk concentration of water. Minimizing free energy with respect to P(α ) yields
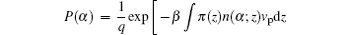
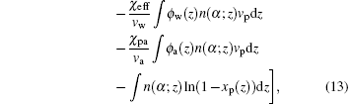
where q is a normalization constant ensuring ∑ α P(α ) = 1, and n(α ; z)dz is the number of ssDNA segments located at z when as ssDNA chain has the conformation α . The volume fraction of air molecules is given by
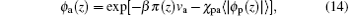
and the volume fraction of water– vapor molecules ϕ w(z) is given by
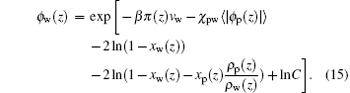
Then equations (6a) and (6b) will be rewritten as
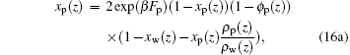 | ) |
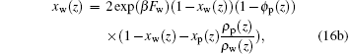 |
respectively. From Eqs. (16a) and (16b) it follows that the difference between local fractions of xp(z) and xw(z) originates from the difference between the free-energy associated with formations of a single ssDNA– water hydrogen bond and the free-energy associated with a single water– water hydrogen bond.[32, 33] The unknowns in the above equations are the position-dependent repulsive fields and the fraction of hydrogen-bonded moieties. The values of these quantities can be obtained by substituting Eqs. (12)– (16) into the packing constraints of Eq. (11). A detailed numerical methodology can be found in Refs. [29], [33], and [35].
In this section, we present some representative results of anomalous switching of ssDNA monolayers.
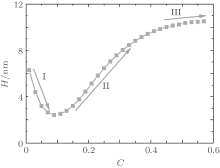
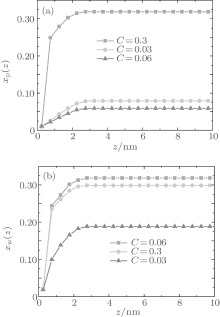
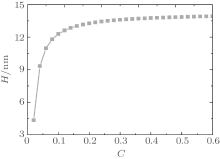
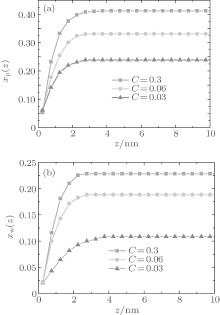
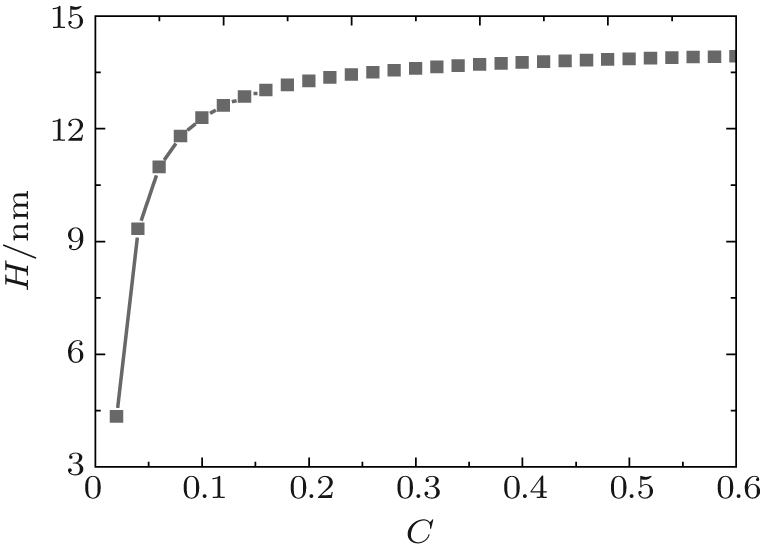
Figure 1 shows the relationship between the average volume fraction of grafted ssDNA chain and water– vapor concentration. From Fig. 1, it is easy to see that the grafted ssDNA chains show anomalous switching as the concentration of water– vapor outside it increases. The height of grafted ssDNA monolayer H is defined as H = ∫ ϕ p(z)zdz/ ∫ ϕ p(z)dz, which measures the amount of stretching of grafted ssDNA chains. In the same way, we show the relationship between the height of grafted ssDNA monolayers and water– vapor concentration in Fig. 2, from which the grafted ssDNA first collapses (in stage I) and then stretches with the increasing water– vapor concentration (in stage II). In other words, a strong dependence of height on water– vapor concentration is obvious. Indeed, in the experiments previously reported, [22] the ssDNA chains are observed to collapse rapidly with the increasing humidity, and then to stretch with a further increase in the water– vapor concentration. In order to understand the origin of this behavior, we investigate variations in hydrogen-bond fraction, and produce the diagrams of local fractions of ssDNA– water and water– water hydrogen bonds each as a function of the distance from substrate surface during the hydration stage as shown in Figs. 3(a) and 3(b). One can see that both ssDNA– water and water– water hydrogen bonds near the monolayer surface are higher at different water vapor concentrations. Interestingly, a similar phenomenon is found within the polyethylene oxide (PEO) monolayer where the density of water is significantly lower than that of bulk water[36]. Weiss et al.[37] found that the connectivity of water– water hydrogen bonds near substrate surface is higher. Similar distributions of local fractions of ssDNA– water and water– water hydrogen bonds at different water– vapor concentrations are found in Fig. 3. This distribution indicates that the ssDNA– water and water– water hydrogen bonds are position-dependent and it also indicates the formation of an intricate hydrogen-bond network.
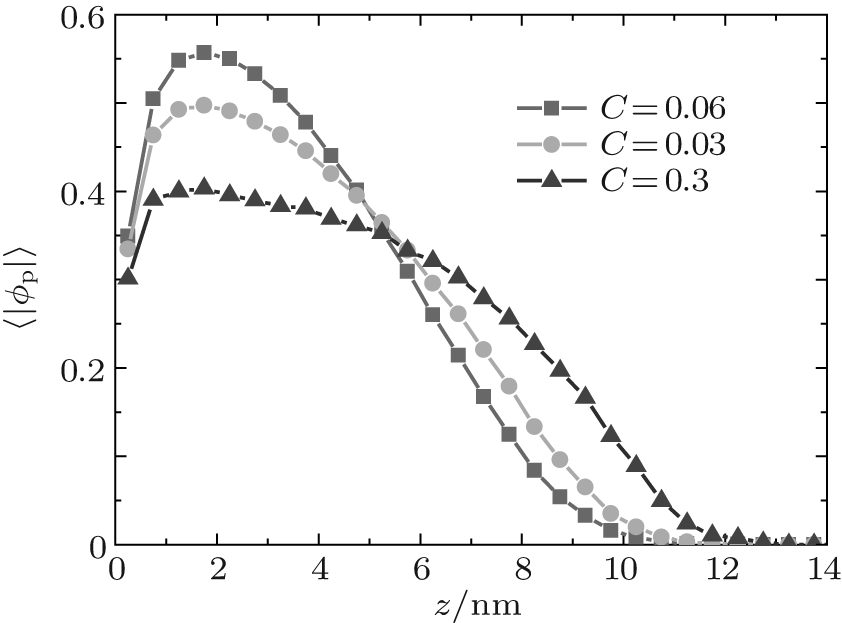 | Fig. 1. Plots of average volume fraction of grafted ssDNA chain versus the distance from substrate surface during the hydration stage. The surface coverage is σ = 0.1 nm− 2. |
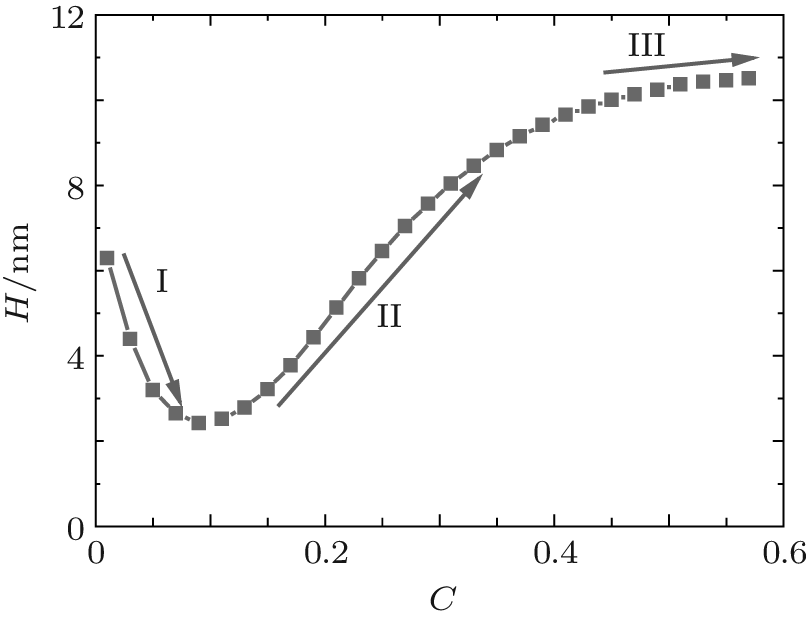 | Fig. 2. Plot of the height of grafted ssDNA monolayer with water– vapor concentration during the hydration stage. |
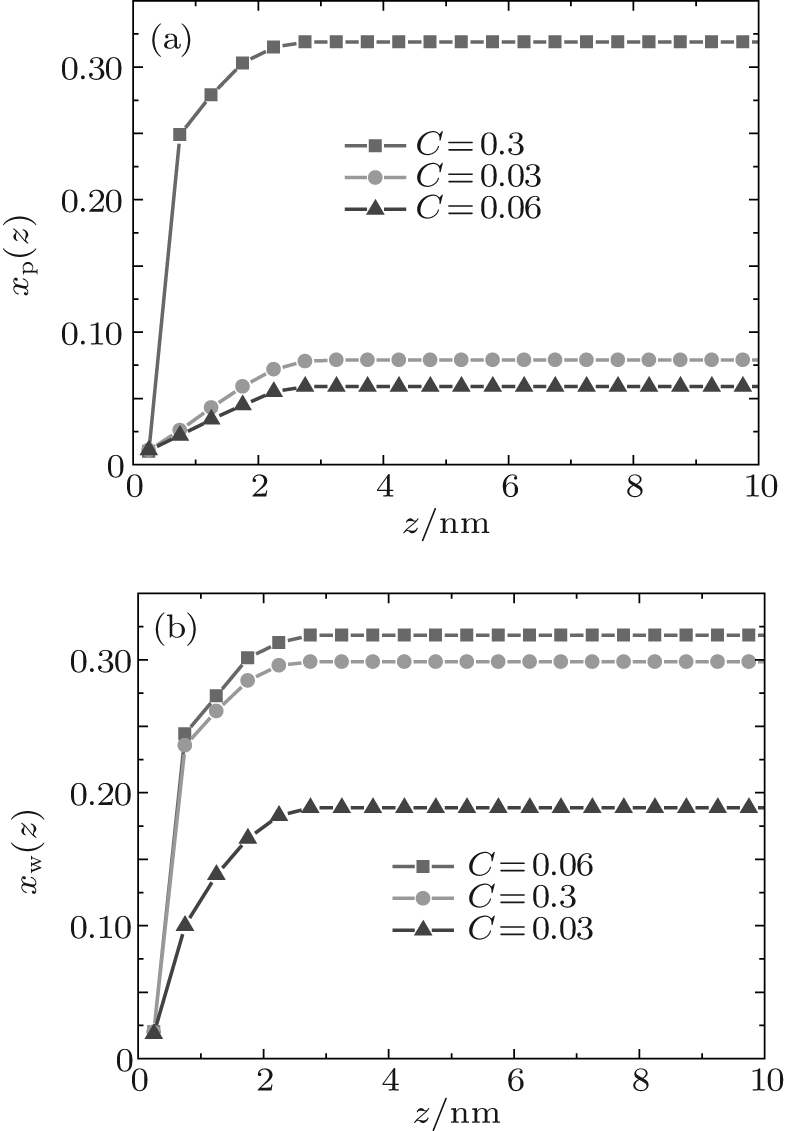 | Fig. 3. Local fractions of hydrogen bonds each as a function of the distance from substrate surface during the hydration stage, for (a) ssDNA– water and (b) water– water hydrogen bonds. |
In order to describe overall behaviors of ssDNA monolayers, we define an average fraction of ssDNA– water hydrogen bonds 〈 | xp(z)| 〉 = ∫ ϕ p(z)xp(z) dz / ∫ ϕ p(z) dz and an average fraction of water– water hydrogen bonds, 〈 | xw(z)| 〉 = ∫ ϕ w(z)xw(z) dz / ∫ ϕ w(z)dz.
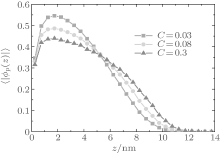
Figure 4 shows the average fractions of ssDNA– water and water– water hydrogen bonds, each as a function of water– vapor concentration. From Fig. 4, it is apparent that the average fraction of ssDNA– water hydrogen bonds first decreases slightly, and then increases abruptly with increasing the water– vapor concentration, while the average fraction of water– water hydrogen bonds first increases rapidly and then decreases slowly. The main reason for the variations in Fig. 4 is presented as follows. Since the initial hydration stage (stage I) is linked to the formation of DNA hydration shells, where water molecules are bound up with ssDNA chains through hydrogen bonds, and an increase in water– water hydrogen bonds leads to the formation of water molecules mediated by a physical network of ssDNA chains, along which water molecule-mediated monomer– monomer attractive interactions are produced. As water– vapor concentrations exceeds 0.15∼ 0.3 (stage II), the ssDNA– water hydrogen bonds increase rapidly, which leads to an increase in repulsive interactions along ssDNA chains. Increasing the ssDNA– water hydrogen bonds leads to a free energy gain, which is balanced by the loss of conformational entropy of chains due to chain stretching, and leads to an increase in the number of intermolecular repulsions. This additional cost of free energy further hinders water– water association, which can be illustrated by the fact that 〈 | xw(z)| 〉 decreases with the increasing water– vapor concentration. Finally, in the third regime of water adsorption (stage III), a small slope of height curves found at water– vapor concentration C = 0.50 suggests that the ssDNA– water hydrogen bonds could be filled, which would cause ssDNA chains to stretch. It is noteworthy that as water– vapor concentration increases, the swelling anomaly is caused directly by ssDNA– water and water– water hydrogen bonds, according to our model. In the whole process of dehydration, we take the values of two parameters: kp = 0.35 and kx = 0.25. This immediately suggests that ssDNA chains should collapse monotonically with the reducing humidity (see Figs. 5 and 6). Our results are consistent with experimental observations.[22]
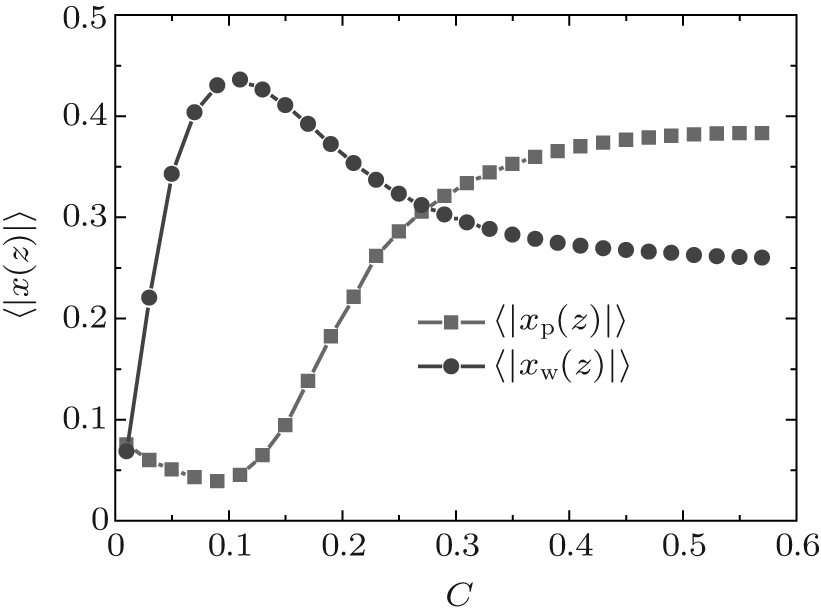 | Fig. 4. Average fractions of ssDNA– water (solid squares) and water– water (solid circles) hydrogen bonds each as a function of water– vapor concentration during the hydration stage. |
 | Fig. 5. Average volume fractions of grafted ssDNA chains each as a function of the distance from substrate surface during dehydration. The surface coverage is σ = 0.1 nm− 2 |
 | Fig. 6. Height of the grafted layer as a function of the water– vapor concentration during dehydration. |
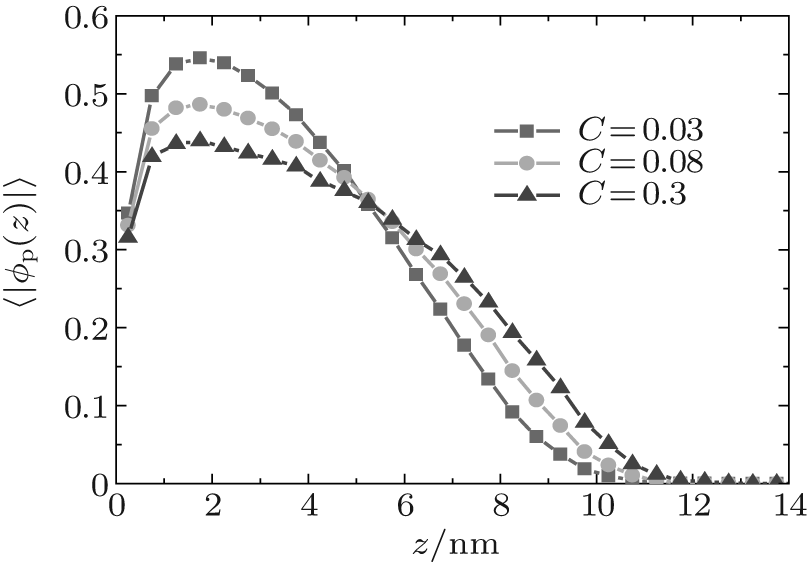
During dehydration, hydrogen bonding decreases with the reducing water– vapor concentration, as shown in Figs. 7 and 8. Decreases in fractions of ssDNA– water and water– water hydrogen bonds indicate that the hydrogen-bond network is disrupted by reducing the water– vapor concentration. It is only at very low water– vapor concentrations (C = 0.02). Notably, the disruption of the hydrogen-bond network occurs only at a very low water– vapor concentrations because there are not enough water molecules available to maintain hydrogen bonding. In order to reduce the free energy, the ssDNA chain subsequently returns to its initial state, accompanied by a complete decrease in hydrogen bonding.
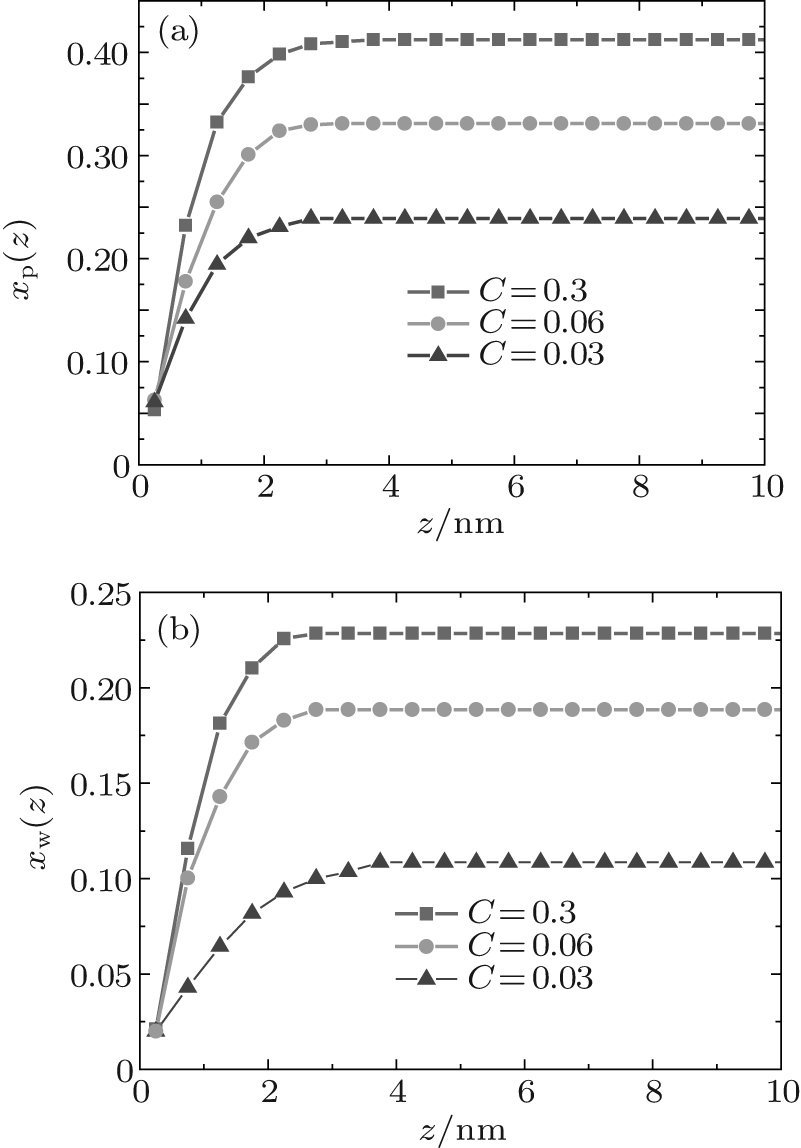 | Fig. 7. Local fractions of (a) ssDNA– water and (b) water– water hydrogen bonds each as a function of the distance from substrate surface during dehydration. |
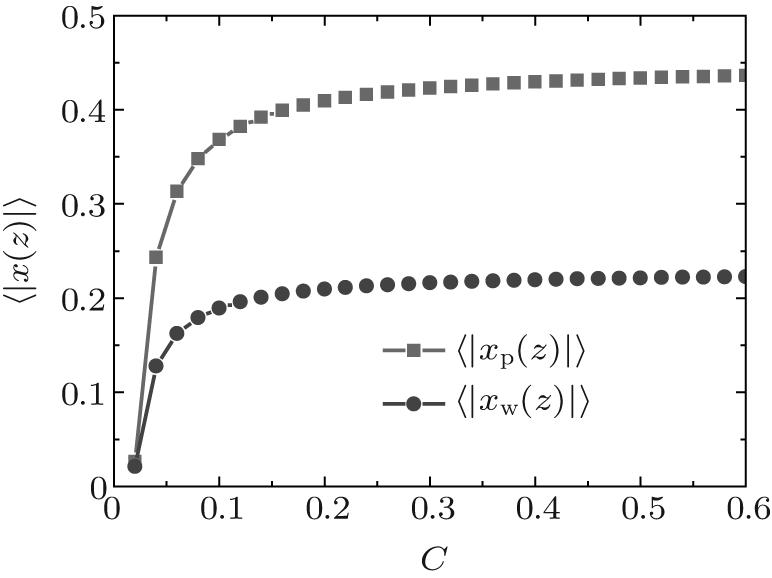 | Fig. 8. Average fractions of ssDNA– water hydrogen bonds (solid squares) and water– water hydrogen bonds each as a function of water– vapor concentration during dehydration. |
By comparing hydration (Fig. 2) with dehydration (Fig. 6), we find that a key signature of the response to ssDNA monolayers is hysteresis in the hydration/dehydration loop. This hysteresis was reported by Mertens et al., [22] experimentally. Bryan[38] explained that there exists a capillary-like force between ssDNA molecules. According to our model, the anomalous switching could be a direct consequence of hydrogen bonding, the capillary-like force could be caused by the effect of hydrogen bonding, and the hysteresis could be attributed to these ssDNA– water and water– water hydrogen bonds that act as the “ cross-linking” points leading to the formation of an intricate hydrogen-bond network. The dissociation occurs only at relatively low water– vapor concentration.
We use a molecular theory to study the anomalous switching of ssDNA monolayers by water vapor. Unlike the method of Wagman et al., [23] we consider both ssDNA– water and water– water hydrogen bonding and their explicit coupling to the ssDNA conformations. Our results show that when at moderate grafting densities, the combination of ssDNA– water hydrogen bonds and water– water hydrogen bonds results in the nonmonotonic dependence of an ssDNA monolayer height on water– vapor concentration: the grafted layer initially shrinks with the increasing humidity until it reaches a minimum, and then increases as humidity increases further. This is in good agreement with the observations by Mettens et al., [22] from which a rather unique (nonmonotonic) switching behavior was revealed when humidity changes. In Ref. [22], the authors also reported a hysteretic behavior: when humidity subsequently decreased, the height of grafted layer decreased monotonically with the decreasing water– vapor concentration. Liu and Zhang[26] also found a similar conformational change of PNIPAM brushes when temperature varies, which can be explained by hydrogen bond effects.[27] We expect that the anomalous switching of ssDNA monolayers and hysteresis will be attributed to the effects of ssDNA– water and water– water hydrogen bonds. Furthermore, we infer that the hysteretic behaviors[22, 26] could occur due to hydrogen bonds effects, and anomalous switching induced by water vapor could be general for a wide range of hydrogen bond polymers (PEO or PNIPAM) monolayers.
According to our theoretical model, we can predict that as humidity in three-dimensional ssDNA monolayers increases, the hybridization between ssDNA– water hydrogen bonds and water– water hydrogen bonds will lead to hydrogen-bond network formation at high grafting densities, and there will be a decrease in the probability of associations between ssDNA– water hydrogen bonds and water– water hydrogen bonds for sufficiently high ssDNA monolayers densities. We also suggest that at a sufficiently high density of ssDNA monolayers the cross-hybridization between ssDNA and ssDNA by hydrogen bonds between complementary base pairs will result in the formation of a cross-linked DNA network. Our predictions could be partly tested by recent experiments on DNA monolayers and concentrated DNA solutions.[39]
The theoretical approach presented in this work has general characteristics which will be applied to other molecular systems, e.g., single-stranded PNA and single-stranded RNA.[22, 40– 43] These molecule systems have great potential for applications in biomaterials. It is worth mentioning that experiments in previous studies were carried out under non-equilibrium conditions. However, we believe that the main anomalous switching effects of ssDNA monolayers by water vapor are captured by our equilibrium approach. Our theoretical results agree well with experimental observations, which supports the suggestion that hydrogen bonding becomes a key element in inducing anomalous switching of ssDNA monolayers by water vapor. Although there certainly exist other contributions (e.g., air molecules), they are unlikely to play a pivotal role in changing the switching behavior of the ssDNA monolayer. The slow non-equilibrium kinetics as observed in the experiments of Ref. [20], can lead to the lateral phase separation, [44] making the monolayer density nonhomogenous. It will be necessary to build a theoretical model that takes into account a three-dimensional non-equilibrium system. Advances in theory, [45– 50] combined with experimental observations, will enable us to modify the designs of biopolymer surfaces for a large variety of applications.
We would like to thank Dr. Ren Chun-Lai for her valuable advice and suggestions.
| 1 |
|
| 2 |
|
| 3 |
|
| 4 |
|
| 5 |
|
| 6 |
|
| 7 |
|
| 8 |
|
| 9 |
|
| 10 |
|
| 11 |
|
| 12 |
|
| 13 |
|
| 14 |
|
| 15 |
|
| 16 |
|
| 17 |
|
| 18 |
|
| 19 |
|
| 20 |
|
| 21 |
|
| 22 |
|
| 23 |
|
| 24 |
|
| 25 |
|
| 26 |
|
| 27 |
|
| 28 |
|
| 29 |
|
| 30 |
|
| 31 |
|
| 32 |
|
| 33 |
|
| 34 |
|
| 35 |
|
| 36 |
|
| 37 |
|
| 38 |
|
| 39 |
|
| 40 |
|
| 41 |
|
| 42 |
|
| 43 |
|
| 44 |
|
| 45 |
|
| 46 |
|
| 47 |
|
| 48 |
|
| 49 |
|
| 50 |
|