
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Two-dimensional arsenic monolayer sheet predicted from first-principles*
[Pu Chun-Yinga) , Ye Xiao-Taob) , Jiang Hua-Longa) , Zhang Fei-Wuc), d) , Lu Zhi-Wena) , He Jun-Baoa) , Zhou Da-Weia)†  ]
]
]
|
|


†Corresponding author. E-mail: zhoudawei@nynu.edu.cn
Projected supported by the Henan Joint Funds of the National Natural Science Foundation of China (Grant Nos. U1304612 and U1404608), the National Natural Science Foundation of China (Grant Nos. 51374132 and 11404175), the Special Fund for Theoretical Physics of China (Grant No. 11247222), and Nanyang Normal University Science Foundation, China (Grant Nos. ZX2012018 and ZX2013019).
Using first-principles calculations, we investigate the two-dimensional arsenic nanosheet isolated from bulk gray arsenic. Its dynamical stability is confirmed by phonon calculations and molecular dynamics analyzing. The arsenic sheet is an indirect band gap semiconductor with a band gap of 2.21 eV in the hybrid HSE06 functional calculations. The valence band maximum (VBM) and the conduction band minimum (CBM) are mainly occupied by the 4p orbitals of arsenic atoms, which is consistent with the partial charge densities of VBM and CBM. The charge density of the VBM G point has the character of a π bond, which originates from p orbitals. Furthermore, tensile and compressive strains are applied in the armchair and zigzag directions, related to the tensile deformations of zigzag and armchair nanotubes, respectively. We find that the ultimate strain in zigzag deformation is 0.13, smaller than 0.18 of armchair deformation. The limit compressive stresses of single-layer arsenic along armchair and zigzag directions are −4.83 GPa and −4.76 GPa with corresponding strains of −0.15 and −0.14, respectively.
Since graphene was isolated from graphite and identified to be stable in an ambient environment by Geim and Novoselov, [1, 2] graphene-like two-dimensional (2D) materials have attracted lots of interest due to their fascinating mechanical, electronic, and transport properties.[3– 5] For example, graphene analogous of two-dimensional boron-nitride (BN) monolayer has been successfully fabricated by a sputtering method and subjected to extensive studies with promising applications in electronics and energy storage.[6– 9] Monolayer sheets of various inorganic layered materials, such as MoS2, WS2, MoTe2, MoSe2, NbTe2, and NbSe2, can be efficiently produced by the liquid exfoliation technique.[10– 13] By using a structural search method in combination with the first-principles calculations, lots of 2D carbon allotropes, nitrogen– graphene alloys, and group III– V compounds have been predicted.[14– 18] These reported 2D layered compounds show different properties ranging from zero gap semimetals to wide gap semiconductors. Similar to graphene, some group-IV elements, such as silicon (Si) and germanium (Ge), have also been predicted to have honeycomb structures, which are named as silicene and germanene.[19, 20] Although monolayers of silicon and germanium are unlikely flat, their electronic structures have much in common with those of graphene. In the experiment, the silicene sheet has already been synthesized by epitaxial growth of Si atoms on a Ag surface or ZrB2 film.[21– 24] Furthermore, a novel 2D boron structure with massless dirac fermions has been predicted and investigated.[25– 27] Recently, the 2D honeycomb network of Ge atoms was successfully fabricated on Pt(111).[28] These studies motivated increasing interest in seeking new nanosheets of other elements. Liu et al.[29] predicted that boron clusters and sheets can be stabilized on the Cu(111) surface, which was confirmed experimentally by Sergeeva et al.[30] In this paper, we are interested in the low dimensional system of As. In fact, gray arsenic (α -As) has a double-layered structure, the bonding between the layers is very weak. If we isolate a 2D arsenic sheet from the gray arsenic, the following issues may be interesting. Is the 2D arsenic sheet stable? What physical and chemical properties will it possess? To address the above issues, we investigate systematically the 2D arsenic sheet using first-principles total energy method based on the density functional theory. The structural character, stability, and electronic properties of the 2D arsenic sheet are investigated in detail. To better understand the mechanical behavior, the strain– stress and compressive curves of the 2D sheet are also discussed. The investigations in this work may provide important information for the potential applications of the 2D arsenic sheet once it is synthesized.
The 2D arsenic sheet was isolated from gray arsenic. To model the single-layer system, there was a 20 Å thick vacuum region between layers to reduce the inter-layer interaction. The structural relaxations were realized by the projector augmented wave (PAW) method[31] implemented in the Vienna ab initio simulation package (VASP).[32] The 4s24p3 electrons were adopted as the valence band. The exchange– correlation energy was treated within the generalized gradient approximation (GGA), using the functional of Perdew, Burke, and Ernzerhof (PBE).[33] In addition, the hybrid HSE06 functional with the screening parameter (ω ) of 0.2 Å − 1 was also employed to calculate the band structures of the 2D arsenic structure.[34] A cutoff energy of 450 eV and a Monkhorst– Pack Brillouin zone sampling grid with a resolution of 2π × 0.03 Å − 1 were used. The dynamic stability of the new structure was examined by the phonon calculations and molecular dynamics. The phonon calculations were carried out with the phonopy code.[35] The first-principles molecular dynamics simulations adopt the constant temperature and volume (NVT) ensemble and were carried out with time steps of 2 fs for a total simulation time of about 4 ps at 1000 K.
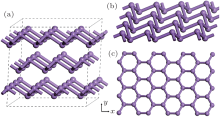
Gray arsenic adopts a double-layered structure consisting of many interlocked ruffled six-membered rings. The bonding between the layers in gray arsenic is weak, thus we may isolate a 2D arsenic sheet from gray arsenic. The final 2D arsenic sheet together with the gray arsenic structure is shown in Fig. 1. Figure 1(b) is a perspective view that displays the puckered configuration of the arsenic sheet with the space group 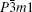
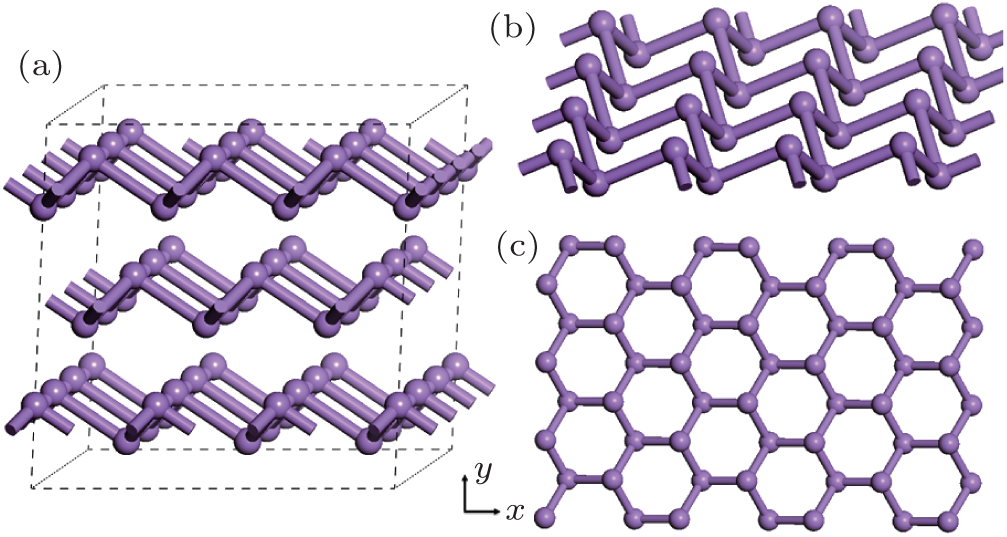 | Fig. 1. (a) A 2 × 2 × 1 gray arsenic supercell, (b) perspective view of 2D arsenic sheet, (c) honeycomb-like structure. |
For the 2D arsenic sheet isolated from the bulk gray arsenic, whether it can stably exist is very important. We first calculated the atomization energy for the arsenic atom in the 2D sheet in order to compare with that in arsenic clusters. The calculated atomization energy is 4.75 eV/atom, which is higher than that in arsenic clusters.[39] The formation energy Ef of single-layer arsenic is a more important indicator and is defined relative to the three-dimensional (3D) bulk ground-state structure as Ef = E2D/N2D − E3D/N3D, where E2D and E3D are the total energies of single-layer and bulk gray arsenic, respectively, [40] and N2D and N3D are the numbers of atoms in the 2D and the 3D unit cells, respectively. The formation energy of the 2D arsenic is calculated to be about 0.021 eV/atom, which is smaller than 0.34 eV/atom of silicene.[41] Such a small formation energy indicates the ease of cleaving a sheet of single-layer arsenic from bulk crystals.
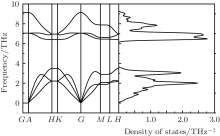
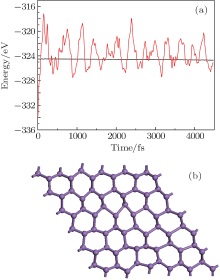
To confirm the dynamic structural stability of the 2D arsenic sheet, we performed the phonon-spectrum lattice dynamics calculations at 0 K using the finite displacement method, as implemented in the Phonopy code. As shown in Fig. 2, no imaginary phonon frequencies are observed in the Brillouin zone for the 2D arsenic structure, indicating the inherent dynamical stability of the arsenic sheet. To further examine the structural stability, we performed the molecular dynamics simulations, which were carried out on a larger 6 × 6 supercell at temperature 1000 K of the Nose thermostat with a step of 2 fs. Figure 3(a) depicts the fluctuations of energy with time during the simulations for the arsenic sheet. It is found that the arsenic sheet is preserved with small fluctuations, which can be relaxed back after full structural optimizations. The structure of the 2D arsenic sheet remains almost intact (see Fig. 3(b)) after 2200 steps. The high thermal stability of the arsenic sheet should be due to the high stability of the strong As– As bond. Therefore, combining with the phonon spectra, we can conclude that the 2D arsenic sheet has dynamical stability.
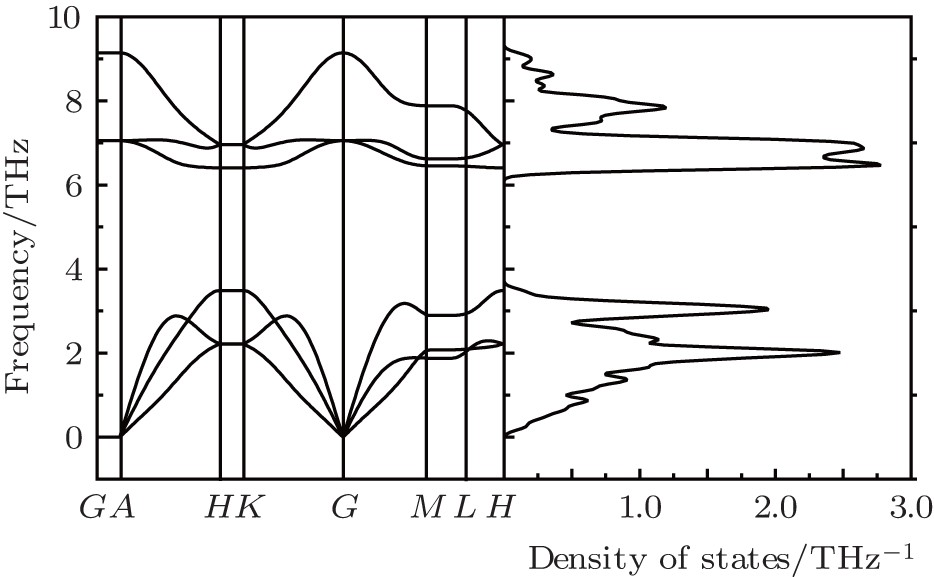 | Fig. 2. Phonon band dispersion curves of arsenic sheet obtained by the finite displacement method. |
It is worth noting that the density functional theory calculation always underestimates the energy gap. To calculate the electronic structure more accurately, we employed a hybrid functional of Heyd– Scuseria– Ernzerhof (HSE06) calculations to regulate the band structures of the arsenic sheet. The electronic band structure of the 2D arsenic sheet is shown in Fig. 4(a). It is found that the valance band maximum (VBM) is located at the G point and the conduction band minimum (CBM) is located in the G– M interval. Distinct from graphene, the 2D arsenic sheet is predicted to be a semiconductor with an indirect energy band gap of 2.21 eV. The band gap is close to that of MoS2 and WS2 monolayers.[42] To gain deeper insight into the electronic structure, we analyzed the total density of states and the orbital-projected atomic density of states of the arsenic sheet in Fig. 4(b). It can be seen that the valence band maximum and the conduction band minimum are mainly contributed from the 4p orbitals of As atoms, which is consistent with the partial charge densities of VBM and CBM in Figs. 4(c) and 4(d). The VBM is composed of the As– As bonding states, and the CBM comes from the anti-bonding states. Thus the charge density of the VBM G point has the character of a π bond, which originates from p orbitals.
 | Fig. 3. (a) The energy fluctuations with respect to time in molecular dynamics simulations at 1000 K. (b) Snapshot of arsenic sheet after 4.5 ps MD simulation at 1000 K. |
 | Fig. 4. (a) Band structures, (b) DOSs, and the partial charge densities for (c) VBM and (d) CBM of arsenic sheet. |
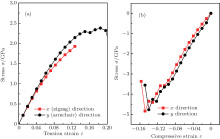
The puckered structure of single-layer arsenic implies that the material may be much more ductile in uniaxial deformation, which would lead to strongly anisotropic mechanical properties. Figure 5 presents the stress– strain curves for uniaxial strains in single-layer arsenic with respect to compression or tension along x and y directions (Fig. 1(c)). Here, the strain is defined as ε = L/L0 − 1, and the stress σ is the Cauchy stress. The x direction is the nearest-neighbor As– As bonding direction, corresponding to pulling a zigzag nanotube of chirality. The y direction is the second-nearest-neighbor direction. Pulling in the y direction corresponds to pulling an armchair nanotube of chirality. When the tensile strain is applied, the ultimate strain in zigzag deformation is 0.13, smaller than 0.18 in armchair deformation. The stress will have a downward trend at large strain, and the maximum Cauchy stress for uniaxial tension in x (zigzag nanotubes) is 1.93 GPa. Compared to being pulled in the x direction, the single arsenic sheet is somewhat stronger in the y direction (relevant for an armchair nanotube), with a maximum Cauchy stress of 2.38 GPa. Thus, we predict the armchair nanotubes to be 2.3% stronger than the zigzag nanotubes and that they can withstand a relatively larger strain. To further investigate the effect of pressure on the arsenic sheet, the compressive stresses along x and y directions were calculated under uniaxial compression. It is found that the limit compressive stresses of single-layer arsenic along x and y directions are − 4.83 GPa and − 4.76 GPa with corresponding strains of − 0.15 and − 0.14, respectively, which means that zigzag nanotubes are more compressive than armchair nanotubes.
 | Fig. 5. Calculated stress-strain curves of arsenic sheet in various tensile and compressive deformation directions. |
We proposed a two-dimensional arsenic nanosheet isolated from bulk gray arsenic. Its dynamical stability is confirmed by means of phonon calculations and molecular dynamics simulations. The 2D arsenic sheet is found to be a semiconductor with an indirect band gap of 2.21 eV. The VBM has π character that originates from arsenic 4p orbitals. The maximum Cauchy stress for uniaxial tension along the x direction is 1.93 GPa, whereas it is 2.38 GPa along the y direction. The compressive stresses along x and y directions were calculated under uniaxial compression. The limit compressive stresses of the single-layer arsenic along x and y directions are − 4.83 GPa and − 4.76 GPa with corresponding strains of − 0.15 and − 0.14, respectively, revealing that the zigzag nanotubes are more compressive than the armchair nanotubes. We hope our results in this paper can provide some guidelines to synthetize the 2D arsenic sheet in experiment.
| 1 |
|
| 2 |
|
| 3 |
|
| 4 |
|
| 5 |
|
| 6 |
|
| 7 |
|
| 8 |
|
| 9 |
|
| 10 |
|
| 11 |
|
| 12 |
|
| 13 |
|
| 14 |
|
| 15 |
|
| 16 |
|
| 17 |
|
| 18 |
|
| 19 |
|
| 20 |
|
| 21 |
|
| 22 |
|
| 23 |
|
| 24 |
|
| 25 |
|
| 26 |
|
| 27 |
|
| 28 |
|
| 29 |
|
| 30 |
|
| 31 |
|
| 32 |
|
| 33 |
|
| 34 |
|
| 35 |
|
| 36 |
|
| 37 |
|
| 38 |
|
| 39 |
|
| 40 |
|
| 41 |
|
| 42 |
|