
{kind=link}
{kind=link}
{kind=link}
First-principles study of structure and nonlinear optical properties of CdHg(SCN)4 crystal*
[Zhang Penga) , Kong Chui-Ganga) , Zheng Chaoa) , Wang Xin-Qiangb) , Ma Yuea) , Feng Jin-Boa) , Jiao Yu-Qiua) , Lu Gui-Wua)†  ]
]
]
|
|


Corresponding author. E-mail: lugw@cup.edu.cn
Project supported by the National Natural Science Foundation of China (Grant No. 51372140), the Youth Scientist Fund of Shandong Province, China (Grant No. BS2011CL025), and the Basic Discipline Research Fund of China University of Petroleum, Beijing, China (Grant No. 01JB0169).
The geometric structure, electronic structure, and optical properties of CdHg(SCN)4 crystal are calculated by using the density functional perturbation theory and Green function screening Coulomb interaction approximation. The band gap of CdHg(SCN)4 crystal is calculated to be 3.198 eV, which is in good agreement with the experimental value 3.265 eV. The calculated second-order nonlinear optical coefficients are d14 = 1.2906 pm/V and d15 = 5.0928 pm/V, which are in agreement with the experimental results ( d14 = (1.4 ± 0.6)pm/V and d15 = (6.0 ± 0.9 pm/V). Moreover, it is found that the contribution to the valence band mainly comes from Cd-4d, Hg-5d states, and the contributions to the valence band top and the conduction band bottom predominantly come from C-2p, N-2p, and S-3p states. The second-order nonlinear optical effect of CdHg(SCN)4 crystal results mainly from the internal electronic transition of (SCN)−.
Nonlinear optical (NLO) crystal materials are widely used in the area of optical computing, optical communications, optical data storage, switching, amplification, and signal processing. In recent decades, a number of NLO crystals with good performances have been synthesized, and their structures and optical properties have been studied. In particular, researchers in Shandong University have synthesized a series of bimetallic coordination compounds ABTC (A = Mg, Ca, Sr, Ba, Mn, Fe, Cu, Zn, Co, Ni, and Cd; B = Co, Zn, Cd, and Hg), and Yuan et al. have studied the crystal structures and the NLO properties of some compounds.[1] The results show that the second harmonic generation (SHG) efficiency of the crystal powder of CdHg(SCN)4 is 13 times larger than that of urea, and CdHg(SCN)4 possesses a good thermal stability and high laser damage threshold. However, the physics mechanism of NLO effect for ABTC crystal is not clear to our knowledge. So a further theoretical study and physical understanding of it are desired.
In theoretical study, the energy band structures are usually calculated in the local density approximation (LDA) or generalized gradient approximation (GGA) based on the density-functional theory (DFT). However, it is known that the band gaps are always underestimated if the calculation is based on the DFT and the calculated band gap generally requires correction, no matter what approximation method is used.[2, 3] There are some methods of correcting the calculations, such as Green function screening Coulomb interaction (GW) approximation, [4– 6] hybrid density-functional theory, [7] self-interaction– correction methods, [8] etc. Among these methods, the GW approximation is regarded as the most reliable. In this paper, the energy band structure of CMTC crystal is computed and their second-order NLO properties are studied with the GW approximation method and the density functional perturbation theory (DFPT).
The GW approximation is based on the set of Hedin's equations that, when solved self-consistently, yield the exact interacting single-particle Green function.[9] In the GW approximation, the vertex corrections are neglected, resulting in a simplified self-energy Σ xc equation, i.e.,
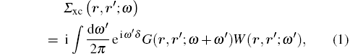
where δ is a positive infinitesimal number, and G and W are the electron Green function and dynamically screened Coulomb interaction, respectively. Hedin’ s equations even in the GW approximation form are still very complicated. In practice, further approximations are introduced, in which both G and W operators are constructed in the quasi-particle approximation by using the Kohn– Sham wave functions ψ n and energies en obtained from the DFT calculations. Therefore, it is called the G0W0 or one-shot GW approximation, where G0 is the electron Green function and is given as
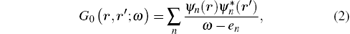
and W0 is the dynamically screened Coulomb interaction

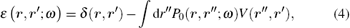
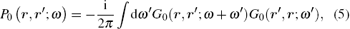
with V being the bare Coulomb interaction, and ε − 1 the inverse dielectric matrix.
The energies ε n are calculated by using the first order perturbation theory, and the perturbation is written as
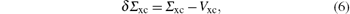
where Vxc is the exchange correlation potential perturbation. The energies
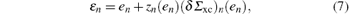
where
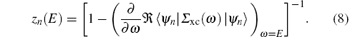
When a light beam transmits through a crystal, the crystal will be polarized by the electric field E. For a weak electric field, the relation between the dielectric polarization P and E is linear. For a strong electric field, however, the linear relation is no longer valid and we can write P as a power series of the electric field as follows:
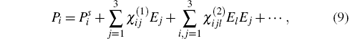
where 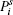
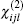
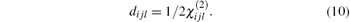
The matrix of dijl consists of 18 tensor elements. According to the symmetry of 
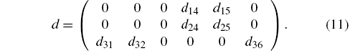
According to Kleinman symmetry, [10] which was devised by Kleinman in 1960, d24 = d31 = d32 = d15 and d36 = d25 = d14. So, only d14 and d15 have nonzero values in the matrix element of dIJ, I = 1, 2, 3; J = 1, 2, 3, 4, 5, 6.
According to the DFPT, [11] we can relate the NLO susceptibilities to a third-order derivative of the energy with respect to an electric field as follows:
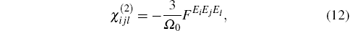
where Ω 0 is the unit volume and 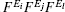
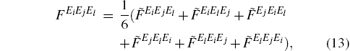
and
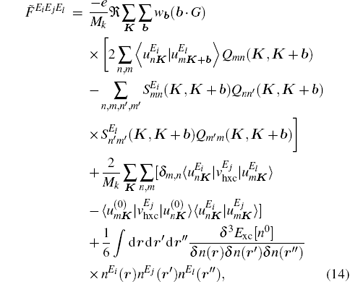
where Mk is the number of k-points, Mk = 4× 4× 4 = 64, Exc is the exchange– correlation energy function, n(r) is the ground-state density, ν hxc is the exchange– correlation potential which is a function of the ground-state density n(r), b is the vector connecting a k-point to one of its nearest neighbours, wb is a weight factor, G is a basis vector of the reciprocal lattice, S is the overlap matrix between Bloch functions at K and K + b, Q is the inverse of the overlap matrix S, and u is the projection of the first-order wave function on the conduction band.
The total energy, electronic structure, band gap, and second-order NLO susceptibilities are calculated using the ABINIT software package.[12] During the calculation, norm-conserving pseudopotential[13] was used, the cut-off energy is 22 Hatree (1 Hatree = 27.2114 eV), and the k-point grid is 4× 4× 8 Monkhorst– Pack[14] in the irreducible Brillouin zone. At first, the structure of CMTC crystal is optimized. The structure is relaxed until the tolerance for absolute difference of total energy below 1.0× 10− 9 Hartree (1 Hartree = 27.2114 eV) is reached. The band number is set to be 400 to guarantee the converge.
The space structure of the CMTC crystal is shown in Fig. 1. The CMTC crystal belongs to a tetragonal crystallographic system, 
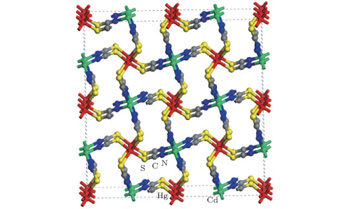 | Fig. 1. Space structure of CMTC crystal. |
| Table 1. Lattice constants and bond lengths of CMTC crystals. |
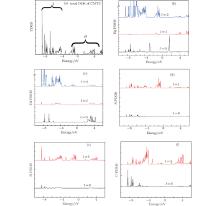
In order to obtain an insight into the contributions to the band structure from Cd, Hg, S, C, and N, the total density of states (TDOS) and partial density of states (PDOS) are computed using a smearing technique and obtained from the eigenvalues, properly weighted at each k point. The calculated TDOS spectrum of the CMTC crystal is shown in Fig. 2(a). The PDOS is computed by using the tetrahedron method. We show the PDOS spectra as well as the angular-momentum projected (l = 0, 1, 2) DOS in Figs. 2(b)– 2(f) in which the orbitals l = 0, 1, 2 represent s, p, and d orbitals, respectively.
 | Fig. 2. (a) TDOS, (b) Hg-PDOS, (c) Cd-PDOS, (d) S-PDOS, (e) N-PDOS, (f) C-PDOS of CMTC crystals. |
The TDOS spectral lines are divided into H and I groups, as shown in Fig. 2(a). By comparing them with PDOS spectral lines of CMTC crystal (Figs. 2(b)– 2(f)), it can be concluded that the peak I (from − 8.57 eV to − 0.41 eV) is for the valence band which is formed mainly from Cd-4d, Hg-5d states, and the valence band top predominantly comes from C-2p, N-2p, and S-3p states. The H group (2.04 eV to 6 eV) is for the conduction band, which is dominantly formed by the C-2p and N-2p states.
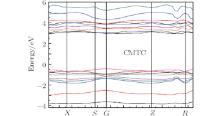
The structure of electronic bands for the CMTC crystal in the vicinity of the forbidden gap is shown in Fig. 3. As can be seen from the figure, the energy band trend is gentler and the bandwidth is small, which is consistent with the complex compound structure of the CMTC crystal. It can be seen that the bottom of conduction band and the top of valence band are both at the same point G in the Brillouin zone. Thus, the CMTC crystal is a direct band gap crystal. The calculated energy gap of CMTC is 3.198 eV, which is close to the experimental value of 3.265 eV.[16]
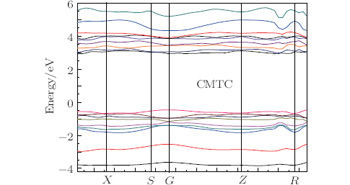 | Fig. 3. Energy band of CMTC crystal. |
Second-order NLO susceptibilities (i.e., SHGs) are studied by using DFPT, and the parameters used are kept the same as the calculations in the GW approximation. Table 2 presents the SHG of CMTC crystal. One can notice from Table 2 that the SHGs of the CMTC crystal are in agreement with the experimental values.
| Table 2. Second-order nonlinear optical susceptibilities of a CMTC crystal. |
The second-order NLO effect of crystal is mainly due to the electronic transition between the valence band top and the conduction band bottom, [18] which indicates that the electron transition in CMTC crystal is the dominant mechanism for SHG. In order to investigate the NLO effect in detail, in this paper the SHG dependence on energy band of CMTC crystal is studied using the energy band structure decomposition method. It is found that occupied states above − 1 eV have a significant effect on the SHG of a CMTC crystal, while the unoccupied states between 3 eV and 6 eV are the main sources for NLO properties. Compared with PDOSs shown in Fig. 2, these occupied states come mainly from N-2p and S-3p orbitals, and the unoccupied states originate mainly from C-2p and N-2p orbitals. Therefore, we can conclude that the SHG effect of CMTC results from the electron transition between the valence band (containing N-2p and S-3p orbitals) and the empty band (containing C-2p and N-2p orbitals).
The structural, electronic, linear, and nonlinear optical properties of a CMTC crystal are studied by the quantum-mechanical calculations based on the GW approximation method and DFPT, and the computed results are in good agreement with the experimental values. Our analysis of the band structure shows that the contributions to the valence band bottom mainly come from Cd-4d, Hg-5d. Moreover, the contributions to the valence band top and the conduction band bottom predominantly come from C-2p, N-2p, and S-3p states. The second-order NLO effects of a CMTC crystal are mainly due to the internal electronic transition in (SCN)− . These conclusions are consistent with the results from the anionic group theory suggested by Cheng Chuang-Tian.[18] The calculated results show that CMTC has good second-order NLO susceptibilities and can be used as a kind of promising crystal material in the NLO field.
| 1 |
|
| 2 |
|
| 3 |
|
| 4 |
|
| 5 |
|
| 6 |
|
| 7 |
|
| 8 |
|
| 9 |
|
| 10 |
|
| 11 | [Cited within:1] |
| 12 |
|
| 13 |
|
| 14 |
|
| 15 |
|
| 16 |
|
| 17 |
|
| 18 |
|