



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Population inversion in fluorescing fragments of super-excited molecules inside an air filament*
Cite this Article
See-Leang Chin, Huai-Liang Xu. Population inversion in fluorescing fragments of super-excited molecules inside an air filament* . Chinese Physics B, 2015, 24(1): 013301 
Permissions
Population inversion in fluorescing fragments of super-excited molecules inside an air filament*
Corresponding author. E-mail: huailiang@jlu.edu.cn
Project supported by the Canada Research Chairs, the Natural Science and Engineering Research Council of Canada (NSERC), the FRQNT, the Canada Foundation for Innovation (CFI), the National Basic Research Program of China (Grant No. 2014CB921300), the National Natural Science Foundation of China (Grant No. 61235003), the Research Fund for the Doctoral Program of Higher Education of China, and the Scientific Research Foundation for Returned Scholars, Ministry of Education of China.
Abstract
An original idea is reviewed. When a molecule is pumped into a super-excited state, one of its decay channels is neutral dissociation. One or more of the neutral fragments will fluoresce. Hence, if a lower state of such fluorescing fragments was populated through other channels but with a lower probability, population inversion of the fluorescing fragments would be naturally realized. This idea seems to be validated, so far, by comparing published work on three hydrocarbon molecules, CH4, C2H2, C2H4, and water vapor, H2O. After super-excitation in a femtosecond laser filament in air mixed with these molecules, the fluorescence from the CH or OH fragments exhibits population inversion, i.e., amplified spontaneous emission was observed in the backscattering direction of the filament.
Keyword:
33.80 Rv; 42.65 Re; 33.20.Xx; femtosecond filament; lasing; super-excited states
1. Introduction
Super-excited states (SES) of molecules whose internal energy is higher than their first ionization potential have been studied extensively after preparing them by extended ultraviolet (XUV) photo-excitation, using synchrotron (XUV) radiation as a light source.[1] It has been proposed that most of such super-excited molecules are prepared in their Rydberg states converging to the ionic ground or excited states of the parent molecules.[1, 2] However, so far, no experiment has been performed to measure the lifetimes of such SESs prepared by XUV photoexcitation.
On the other hand, the SESs of molecules can also be prepared using an intense near-infrared femtosecond laser pulse such as a Ti-sapphire 800-nm femtosecond laser pulse through multiphoton absorption processes.[3] In this case, after excitation by intense visible or near-infrared femtosecond laser pulses, super excited states can be prepared as a coherent superposition of a number of electronic states having different electronic configurations embedded in the ionization continuum.
When a molecule is pumped into a super-excited state, one of its decay channels is neutral dissociation, which leads to fluorescing fragments. Meanwhile, it is also probable that the same fragment could be produced at a lower level via other decay channels. Therefore, in the case of the lower state of such fragments being populated with a lower probability, population inversion of the fragments would be naturally realized, which means that the medium of the excited fragments constitutes a gain medium, thereby exhibiting amplified spontaneous emission (ASE). Our recent observation of ASE from the fragments generated from hydrocarbon molecules CH4, C2H2, and C2H4 and water vapor molecules H2O inside a laser-induced filament in air is an indication that there is indeed a net population inversion in the fluorescing fragments.[4, 5]
In the past two decades, it has been demonstrated that propagation of high-power femtosecond laser pulses in air leads to a high intensity core at 1013– 1014 W/cm2 due to the nonlinear process of “ femtosecond laser filamentation” , which results from the dynamic balance between Kerr self-focusing and defocusing by the plasma produced by multiphoton/tunnel ionization of air molecules.[6] The intensity is high enough to induce multiphoton excitation of molecules in the air, which enables the occurrence of an interesting phenomenon of “ air lasing” with either N2 or O2 as the gain medium.[7– 12] Air lasing was first demonstrated[7] by one of us (SLC) and his group from the detection of the backscattered fluorescence of nitrogen molecules in a filament, in which it was found that the fluorescence intensity of N2 at 357 nm increases exponentially as the filament length increases. The population inversion in nitrogen ASE lasers was initially ascribed to the recombination of free electrons with ions in the plasma[7] or electron impact.[11, 12] More recently, the mechanism of ASE type lasing in an air filament through the excitation of nitrogen molecules has been better understood.[12– 14] On the other hand, the mechanism for ASE lasing resulting from stimulation of O2 molecules in air by 226-nm and 100-ps laser pulses was ascribed to two-photon dissociation of O2 and the subsequent two-photon resonant excitation of atomic oxygen fragments, which produce the emission at 845 nm.[8]
In the following, we will review two sets of experiments of laser-induced filamentation in gaseous media of air mixed with hydrocarbon molecular species such as CH4, C2H2, C2H4, or H2O vapors, in which ASE from neutral fragments such as CH and OH fragments was observed in the backscattering direction of the filament. The underlying mechanism would be different from that of population-inverted molecular nitrogen and atomic oxygen in air. As a possible scenario, formation of the neutral fragments in excited states prepared after the dissociation of the highly electronically excited (or super-excited) parent molecules prepared in the laser-induced filament is proposed.
2. Experimental observation of gain
In this section, we first provide experimental observations of ASE measured in the fluorescence of the neutral fragments generated from CH4, C2H2, and C2H4 mixed in air at one atmospheric pressure when filamentation of a 50-fs Ti-sapphire laser occurs in the mixture. Fluorescence in the back-scattering direction was measured to show the gain, because in the forward direction, the pump light is too strong to be easily filtered out.
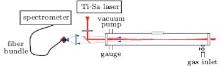
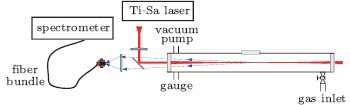 | Fig. 1. Experimental setup for measuring backscattered fluorescence gain in three mixture gases: 2% CH4, 2% C2H2, or 2% C2H4 in air at 1 atm. We detected the CH signal at around 430 nm. The CPA was a Ti-sapphire laser which delivered 800 nm/50 fs pulses and generated filaments at the center of the gas cell. The backward pointing arrows indicate the backscattered fluorescence light that was collected by a fused silica lens affixed to the fiber head coupled to the spectrometer.[7] |
As shown in Fig. 1, the back-scattered fluorescence was collected in a way similar to that in Ref. [7]. The backward fluorescence was collected with a fused silica lens affixed to the entrance end of a 3-m-long fiber bundle (Princeton Instruments, Model LG-455-020), and guided through an imaging fiber adaptor into the entrance slit of a spectrometer (Acton Research Corporation, Spectra Pro-500i) equipped with an intensified charge-coupled device (ICCD) camera (Princeton Instruments, PIMAX512). In addition, a 2.5-nm narrow band-width interference filter that transmits 431-nm light was placed in front of the fiber head to avoid introducing the strong white light into the fiber. In addition, images of the filament (to measure the filament's length) from the lateral direction of the filament were also recorded.
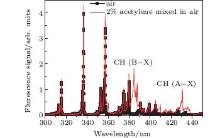
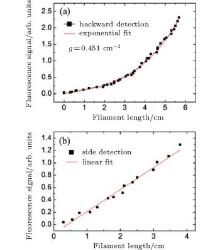
Figure 2 shows a typical spectrum of filament-induced fluorescence from the gas mixture of 2% acetylene (C2H2) in air (red), and that of pure air (black), which was recorded from the side. The fluorescence signal appearing at around 431 nm corresponds to the A2Δ – X2Π (0, 0) transition of CH fragments.[15] All three kinds of hydrocarbons could be fragmented via the excitation through the SES of the molecules inside the filament, resulting in CH fluorescence in each case.[3, 16] The fluorescence spectra for the mixture of air with ethylene (C2H4) and air with methane (CH4) have also been measured. They both show the fluorescence of CH at 431 nm (not shown). We monitored the fluorescence at around 431 nm as an example to examine whether the fluorescence gain is achieved. The back-scattered fluorescence signals plotted as a function of the filament length are shown in Fig. 3. In these figures, it is clearly shown that the fluorescence signals increase nonlinearly as the filament length increases. These curves are fitted well by the gain equation
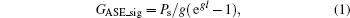 |
in which the gain of ASE, GASE, is expressed using the length l of the gain medium (i.e., the filament length), the gain g, and the spontaneous power Ps. When there is no gain (g = 0), this indicates that the gain of the ASE has been achieved.
 | Fig. 2. Scan of fluorescence spectra from pure air (black) and air contaminated by 2% C2H2 (red). Two CH bands are indicated. We select the CH band around 429– 432 nm and measure the gain (ASE).[4] |
The fluorescence from the fragments CH of these molecules has been recognized to come from super-excitation by the 800-nm femtosecond laser pulse.[3, 16] A qualitative description of this population inversion during strong field super-excitation of molecules is given below.[17, 18]
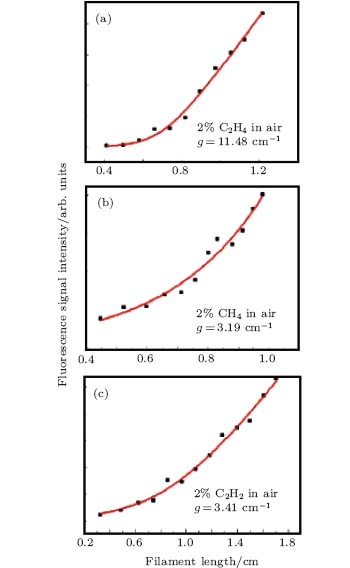 | Fig. 3. Backward integrated fluorescence signal at around 430 nm versus filament length for 2 % of hydrocarbons (a) C2H4, (b) CH4, and (c) C2H2 in air.[4] |
As shown schematically in Fig. 4, a molecule is excited from the ground state into the super-excited state via multiphoton absorption (a series of upward pointing solid arrows), similar to what occurs in XUV or VUV excitation with one-photon absorption (the long upward solid arrow) — a group of the Rydberg states would be in resonance, converging to the ionization limit of an excited or ground state ion.[1, 2] However, to make possible the excitation of molecules to SES by multiphoton absorption, interference stabilization or population trapping may have to occur in order to leave behind a significant population in the Rydberg states, or super-excited states.[19] Briefly, an extra photon is absorbed (upward pointing dashed arrow on top of the solid arrows) exciting the molecule from the resonant super-excited states (Rydberg states) into the continuum. The strong laser field would instantaneously couple the continuum with some neighboring Rydberg states (dashed downward slanting arrow). These super-excited states would further absorb one photon into the continuum (independent upward pointing dashed arrow). The two one-photon transitions from different super-excited states into the continuum (the two upward pointing dashed arrows) would interfere with each other. Part of the interference would be destructive interference, and thus would leave behind some population in the super-excited states. For details, see Ref. [19]. Physically speaking, because of the broadband nature of the pump femtosecond laser pulse, many neighboring Rydberg states would be excited, resulting in an excited wave packet.
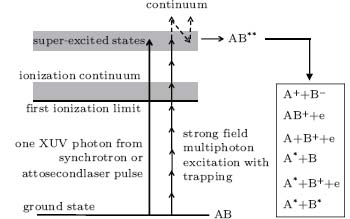 | Fig. 4. Schematic of the excitation of a molecule by either single XUV photon absorption or by multiphoton absorption in an intense laser field with population trapped in the SES. The parent molecule is denoted by AB, where A and B could each be either an atom or a molecule. The super-excited state is denoted by AB**. Its rapid fragmentations into various fragments via different pathways are given in the panel at the right-hand side. |
The super-excited parent molecule denoted as AB** would decay via several possible channels as shown in the right hand panel in Fig. 4. Fluorescence would occur in the last three channels. If population inversion of the fragments could be realized, the fragments would act as a gain medium for a seed pulse with an appropriate wavelength. Otherwise, the fluorescence would exhibit ASE type lasing action if the medium is long enough.
In the following we will concentrate on our study of the fluorescence gain from the laser induced filaments obtained when methane is used as a sample gas. For the dissociation of methane molecules in the intense laser field, the appearance of the product of CH (A2Δ ) indicates that the parent molecule has been excited to the SES by the intense laser according to quantum chemistry calculations[20, 21] and experimental investigations.[22– 24] The dissociation channels would be even more numerous, since some dark products in their ground states, which are not visible, might also be produced, but the ASE action of the CH (A2Δ ) at 431 nm clearly indicates that the SES neutral fragmentation channels dominate.
Next, we will discuss an experimental observation of ASE measured in the fluorescence of hydrogen monoxide (OH) fragmented from water molecules.[5] The experiments were done by using a 12 mJ/50 fs, 10 Hz Ti:sapphire laser beam, as shown in Fig. 5(a). A 5-cm-long filament was created in air over a beaker partially filled with distilled water, leaving an open area above the water in order to create an environment with a water vapor concentration of about 2% at room temperature (22 ° C).[25] The backscattered fluorescence from the filament zone was collected with a fused silica lens and sent to an ICCD gated spectrometer (Acton Research Corporation, Spectra Pro-500i) through a fiber coupler. The fluorescence from the side was obtained by imaging the filament onto the entrance slit of the ICCD gated spectrometer using two identical fused-silica lenses and a periscope.
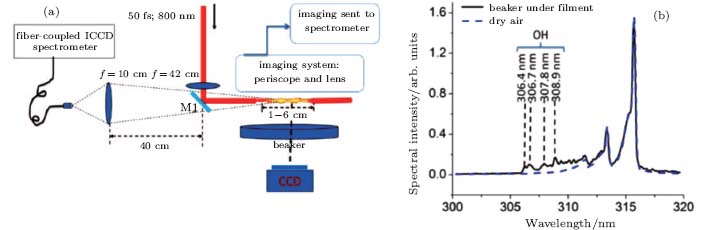 | Fig. 5. (a) Schematic of ASE experiment setup for water vapor measurement and (b) typical spectrum in the range of 300– 320 nm for filament-induced fluorescence of water vapor in air.[5] |
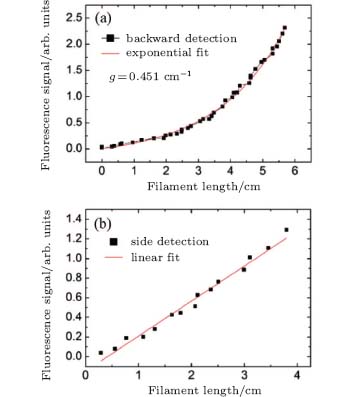 | Fig. 6. The fluorescence intensity of OH at 308.9 nm versus the filament length. The fluorescence is recorded in backward (a) and lateral (b) direction.[5] |
Fluorescence coming from the water vapor was found in the range of 306– 309 nm, as shown by the black curve in Fig. 5(b), at room temperature (22 ° C) and with a pump pulse energy of 2 mJ. The fluorescence was identified as OH* radiation from A2Σ – X2Π . The fluorescence around the peak of 315 nm is from molecular N2. The background spectrum without the water vapor was also detected, as shown by the blue dashed curve in Fig. 5(b), which shows that neither N2 or O2 molecules in air contribute to the fluorescence of OH at around 308.9 nm. The spectral peak intensities of 308.9 nm from OH in the backward and lateral directions were measured under different input pump pulse energy, which can be converted to different filament length.[7] The results are shown in Fig. 6(a) (detected from the backward direction) and Fig. 6(b) (detected from the side). In Fig. 6(a), the backscattered OH fluorescence increases exponentially as the filament length increases, while in Fig. 6(b), the OH fluorescence measured from the side has a linear dependence with the filament length. If the OH fluorescence signal is incoherent, the OH fluorescence emission detected in the backward and lateral directions should give similar results. However, in our experiment, the on-axis backward OH fluorescence signal increases much faster (increases exponentially) with the filament length than the one detected from the side (which increases linearly). This indicates the existence of ASE.
The lasing action is also linked to the SES dissociation process of H2O molecules in intense laser fields, as shown by the energy diagram in Fig. 7, in which 12.6 eV is indicated as the ionization potential (IP) of H2O molecules. Two distinct dissociation channels for H2O molecules below the ionization potential are related to OH generation.[26, 27] One is the channel of dissociating into H(2S) + OH (X2Π ), giving rise to H and OH fragments in the ground states.[27] The other is the channel of dissociating into H(2S) + OH(A2Σ + ), resulting in H in the ground state and OH in the excited state.[26] The threshold energy of 9.136 eV for exciting this channel comes from Refs. [26]– [28], where single vacuum ultraviolet (VUV) photons were used in the super-excitation (below the ionization threshold) and dissociation of H2O. The OH* fluorescence at 306– 309 nm in this experiment was identified as the transition from A2Σ + – X2Π . If the dissociation channels above the ionization potential (12.6 eV) were possible, the excited state of H, namely H*, would be generated together with OH*(A) (see Fig. 7).[29] However, during the experiment, no spectral line from the Balmer series corresponding to H* radiation was observed. Thus, it can be assumed that inside the filament the water vapor molecules were dissociated through the channel H (2S) + OH (A2Σ + ). The transition in OH (A2Σ + – X2Π ) emits fluorescence in the range of 306– 309 nm, with a peak at 308.9 nm. However, if other, lower lying states existed that contributed to the channel H2O → H(2S) + OH(X2Π ), this would reduce the population inversion in the ensemble of the OH population. Evidently, this latter channel is less probable, because gain was observed.
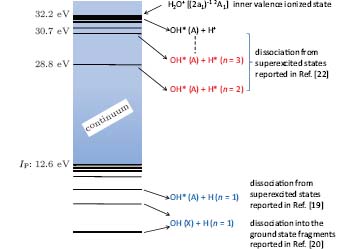 | Fig. 7. Schematic energy diagram for molecular H2O and its possible dissociation channels.[5] |
The density of states is very high in the energy range around 9 eV according to Refs. [26]– [28]. These states were considered to be super-excited states below the ionization threshold. Under strong broadband femtosecond laser field excitation, these states were strongly coupled through the absorption of 6 photons (∼ 9 eV) of the Ti-sapphire laser pulse at the central wavelength of 800 nm. In the experiment, the strong field was set by intensity clamping in the air filament where the intensity was of the order of 5 × 1013 W/cm2. This intensity is sufficient to ionize nitrogen molecules through 8-photon absorption.[7] It is thus sufficient to excite and even saturate the super-excited states of H2O through 6-photon absorption. This super-excited state would decay in the channel H (2S) + OH (A2Σ + ). On the other hand, because of the lack of resonance, the excitation of the channel H2O→ H(2S) + OH(X2Π ) is less probable. Consequently, more excited OH radicals were generated, resulting in the observation of ASE or population inversion in OH*.
3. Decay of super-excited states
Super-excited states have a short lifetime because of the inherent broadband nature of the super-excited state and its coupling to the continuum. As shown in Fig. 4, a super-excited state as defined above can be reached either by the absorption of one XUV photon (from synchrotron radiation, for example)[1, 2] or by multiphoton absorption in a strong laser field through population trapping.[17– 19] Such a super-excited state, being in the ionization continuum, would interact with the latter making its lifetime very short. So far, we have measured that the lifetime is of the order of 100 fs, by using a pump– probe technique for a few molecules (O2, NO, CH4, H2).[30– 33] In the following, we review an experimental example for the lifetime measurement of SES in the fluorescence of CH fragmented from methane molecules.[30]
To measure the lifetime, the experiment was done using a pump– probe method, with the methane molecules being super-excited by an intense femtosecond 800-nm Ti-sapphire laser pulse and probed by its second harmonic. A Ti-sapphire laser system (Spectra Physics Tsunami/Spitfire) delivered slightly negative chirped 60 fs (transform limited pulse: 42 fs), 1 kHz, 2 mJ pulses. The laser beam was then separated into two arms by a 50/50 beam splitter. One was used as the pump beam (∼ 900 μ J/pulse). The other was sent to an OPA to generate the infrared probe pulses at 1338 nm with a pulse duration of about 50 fs (FWHM) and a pulse energy around 90 μ J. A delay line was used to control the delay time between the two pulses. Both of the laser beams were focused by a plano-convex lens (f = 30 cm) into a vacuum chamber filled with pure methane (CH4 Praxair) at 20 Torrs. The fluorescence signal was recorded by a gated PMT (Hamamatsu R5916U-52).
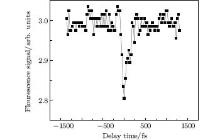
The fluorescence of CH fragmented from methane molecules via SESs was discussed in the last section. In this experiment, the fluorescence signal of the CH (A2Δ → X2Π ) band at 431 nm was measured as a function of the delay time between the pump and probe pulses as shown in Fig. 8. The fluorescence signals were integrated within the wavelength range from 428.5 to 433.5 nm, as well as within a time interval of 600 ns, in order to add up all the fluorescence in the temporal domain. It can be clearly seen from Fig. 8 that when the delay time between the pump and probe is around zero, an obvious decrease of the fluorescence signal takes place. The decrease in fluorescence occurs within a dip width of about 160 fs (FWHM). Since the maximum correlation time of the pump and probe pulses is less than the dip's temporal width, the possibility of cross correlation of two pulses is excluded. Note that outside the dip the fluorescence signals remain almost constant, i.e., the probe pulse does not affect the dissociation any more. This observation clearly indicates that the dissociation channel of the CH excited fluorescing state has an intermediate state with a lifetime of less than 160 fs, which can be destroyed by the present probe pulse.
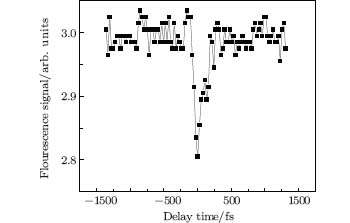 | Fig. 8. Fluorescence signal of CH at around 431 nm versus the delay time between the pump and probe pulses; negative delay time means the probe pulses occur after the pump pulses.[30] |
The decrease in fluorescence signal is slightly greater than 5 percents of the signal obtained when the probe beam does not temporally overlap the pump pulse. This may mean that the probability of SES being destroyed by the present probe pulse is small, or that other channels exist with intermediate states that are not destroyed by the present probe. If an SES has a small probability of being destroyed, it would mean that the state is really imbedded inside the ionization continuum. This is because the probe pulse would not be able to efficiently couple an SES state inside the continuum to another state in the continuum such as ionization, etc. We emphasize that the fluorescence depletion could not be explained by a simple ionization scheme wherein the neutral CH products are generated by disintegration of the CH4+ ion or generated by electron– ion recombination, since the lifetimes of these two processes are very long, in the nanosecond timescale, so we would not have observed the depletion of the fluorescence only in a time zone of around 160 fs. Similarly, the fluorescence depletion does not refer to the direct interaction between the probe laser pulse and CH2 or CH (A) species either, because the depletion takes place immediately after the first excitation without any time delay, while the fluorescence lifetime of CH (A) is normally in the nanosecond timescale.
4. Discussion and conclusion
In this paper, we review the excitation and decay of super-excited states of molecules in intense laser fields. The neutral dissociation of super-excited states may lead to the fluorescing fragments. With the observation of lasing actions in the form of ASE[4, 5, 7] for the hydrocarbon molecular species such as CH4, C2H2, C2H4, as well as H2O vapor inside an air filament, the super-excitation of molecules in intense laser fields was discussed. Such a universal phenomenon could be exploited in many applications eventually.
Another type of super-excitation of molecules could be foreseen. That is, an attosecond laser pulse currently realized by high order harmonic generation, because of its very short central wavelength in the XUV, would probably excite a whole new class of super-excited molecules. The super-excitation by the central XUV wavelength could belong to one-, two- or many-photon absorption into the continuum since the intensity of the attosecond laser pulse could be strong. The super-excited states could span a very broad energetic region in the continuum and the lifetime would most probably be sub-femtosecond depending on the intensity of the attosecond laser pulse. The higher the intensity is, the broader the super-excited bandwidth will be because more states would be coupled with the continuum, and thus the lifetime must be shorter. An indication is the super-excitation of nitrogen molecule by the VUV photons of 23.1 eV from high-order harmonics.[34] Using a femtosecond probe pulse, the decay time of neutral dissociation into nitrogen atoms was measured to be less than 25 fs. This value is to be compared with the decay time of ∼ 100 to 200 fs in the molecules O2, NO, CH4, and H2 pumped by the 800 nm/50 fs Ti-sapphire laser pulse in our laboratory.[30– 33]
We acknowledge Prof. Kaoru Yamanouchi of the University of Tokyo for careful reading of the manuscript and stimulating discussion. We thank Prof. Fanao Kong of the Institute of Chemistry, Chinese Academy of Sciences for fruitful discussion on the super-excitation of molecules.
Reference
| 1 |
|
| 2 | [Cited within:3] |
| 3 |
|
| 4 |
|
| 5 |
|
| 6 |
|
| 7 |
|
| 8 |
|
| 9 |
|
| 10 |
|
| 11 |
|
| 12 |
|
| 13 |
|
| 14 |
|
| 15 |
|
| 16 | [Cited within:2] |
| 17 | [Cited within:2] |
| 18 |
|
| 19 |
|
| 20 |
|
| 21 |
|
| 22 |
|
| 23 |
|
| 24 |
|
| 25 |
|
| 26 |
|
| 27 |
|
| 28 |
|
| 29 |
|
| 30 |
|
| 31 |
|
| 32 |
|
| 33 |
|
| 34 |
|