




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Control of electron localization in the dissociation of *
Cite this Article
Jia Zheng-Mao, Zeng Zhi-Nan, Li Ru-Xin, Xu Zhi-Zhan, Deng Yun-Pei. Control of electron localization in the dissociation of * . Chinese Physics B, 2015, 24(1): 013204 
Permissions
Control of electron localization in the dissociation of *
Corresponding author. E-mail: zhinan_zeng@mail.siom.ac.cn
Project supported by the National Natural Science Foundation of China (Grant Nos. 11127901, 60921004, 11134010, 11222439, 11227902, and 61108012) and the National Basic Research Program of China (Grant No. 2011CB808103).
Abstract
The molecular dissociation with a two-laser-pulse scheme is theoretically investigated for the hydrogen molecular ion (
Keyword:
32.80.Rm; 33.80.Rv; 42.50.Hz; 42.65.Ky; dissociation probability; hydrogen molecular ion; terahertz pulse; time-dependent Schrödinger equation
1. Introduction
Diatomic molecules and molecular ions in intense laser fields continue to attract the attention of the atomic and molecular physics community despite several years of study. One of the main goals in this research direction is to find a way to selectively break and form molecular bonds in photochemical reactions.[1] With the advent of new laser technologies, in particular the carrier envelope phase (CEP) stabilized few-cycle pulses and the isolated subfemtosecond pulses, several control strategies have been proposed, taking advantage of controlling the motion of the electron wave packet in atoms and molecules.[2– 9] The hydrogen molecular ion is of special interest since it is the simplest molecule, consisting of three particles only. It plays an important role in the theoretical and experimental studies of electron localization in the dissociation of molecules. In the previous work, [10– 12] coherent control with a two-color laser field to investigate the asymmetry dissociation of HD+ and 
In our previous work, [17, 18] a time-delayed terahertz (THz) pulse was utilized to steer the electron motion of
2. Simulation model and parameters of the electric fields
We use a reduced-dimensional model for the
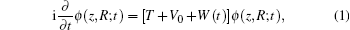 |
where T is the free Hamiltonian of the system, V0 stands for the Coulomb interactions, and W(t) describes the interaction with the external laser field. For our model, the kinetic energy T reads
 |
and the Coulomb potential is
 |
where R is the relative internuclear distance, z is the electronic coordinate with respect to the center of mass of the two nuclei, and μ = (1/mp+ 1/mn)− 1 and μ e = (mp+ mn)/(mp+ mn+ 1) are the reduced masses, with me, mp, and mn being the masses of the electron, the proton, and the second nucleus (me = 1; mp = 1837; mn = 1837, 3674, 5511 for
The interaction with the laser field is written in the dipole approximation (in the length gauge) as
 |
The UV pulse is defined as E2 (t) = E20 sin (π t/T2)2sin (ω 2t), where T2 is the total length of the pulse, i.e., 7.9 fs. The THz pulse is defined with the vector potential A(t) = − E10/ω 1 sin (π t/T1)2cos (ω 1t), where T1 is the total length of the THz pulse; then E1 (t) = − ∂ A(t)/∂ t. The total simulation time is defined by tend, and Δ t is the time delay between the two pulses, which is defined as the difference between the envelope peaks of the two electric fields. When the envelope peak of the THz pulse is ahead, Δ t is positive, otherwise, Δ t is negative. The two channels of the dissociation of these three molecule ions are defined as[9, 13, 16, 31]
 |
where Rmax corresponds to the boundary of the R axis, and φ (z, R; tend) is the final wave function of our system. In the simulation, tend = 106.8 fs when P± , the probabilities of the electron localized on one of the protons (left − or right + ), are stable. The electron localization starts from about R = 5.7 a.u. So, we use R0 = 5.7 a.u. in the definition of P± .[17]
In this simulation, the linearly polarized 228-nm UV pulse with an intensity of 5.0× 1014 W/cm2 and a pulse length T2 of 7.9 fs is used to resonantly excite the electron wave packet onto the dissociative state 2pσ u. Then the 25.6-μ m (11.7 THz in frequency) THz pulse with an intensity of 3.8× 1012 W/cm2 and a pulse length T1 of 85.3 fs is used to steer the electron motion.[32] This THz pulse does not induce any further ionization. In the calculation, we set the dissociation asymmetry parameter as A = (P+ − P− )/(P+ + P− ).
3. Simulation results and discussion
To reveal the details of the electron localization control of
 |
In the definition of PD± , we also choose R = 5.7 a.u., as depicted in Fig. 1(a). Figure 1(b) shows the temporal evolution probabilities of different parts of
 | Fig. 1. (a) Time-dependent electron probabilities of different parts of |
In Fig. 2, we present the probabilities of electrons localized at the right (proton side, cycles) and the left (second nucleus side, stars) z axis, for the interaction of
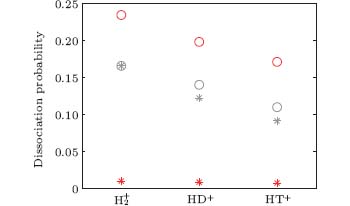 | Fig. 2. Dissociation probabilities with electron localization along the right (proton side, cycles) and the left (second nucleus side, stars) z axis for the interaction of |
 | Fig. 3. (a) Asymmetry parameter A and ionization probability I as functions of Δ t with I1 = 3.8× 1012 W/cm2 and I2 = 5.0× 1014 W/cm2 for |
Figure 3(b) depicts the dissociation asymmetry parameter A and the ionization probability I of
4. Simulation with the coupled equations
But why is the THz pulse useful? As shown in Refs. [17] and [18], the electron localization by using two laser pulses of UV and THz spectral regimes can be much higher than that in the previous work[13] because there is an effective time for controlling the molecular dissociation. The half period of the control electric field needs to match the effective time of the molecule dissociation if a high control ratio is achieved. For
 |
where Vg (R) and Vu (R) are the binding and the dissociative potentials, respectively. 

 | Fig. 4. (a) Calculated results with the coupled equations (7). Asymmetry parameter A of |
Figure 5 shows the largest probability of electrons on the ψ r proton (the right nucleus) by adjusting the time delay dt between the pre-excitation of the Gaussian-type wave packet and the envelope peak of the THz pulse (25.6-μ m), with the nuclear mass of the molecule M = 2.0mp, 16.0mp, 32.0mp.[18] For
 | Fig. 5. Calculated results of the probabilities of electrons on states ψ g, ψ u, ψ l, and ψ r with the coupled equations (7). The mass of the molecule and the time delay between the pre-excited Gaussian-type wave packet and the THz pulse are (a) M = 2.0mp, dt = 49.2 fs; (b) M = 16.0mp, dt = 28.5 fs; and (c) M = 32.0mp, dt = 10.6 fs. (d) The THz pulse utilized throughout this paper. |
5. Summary
We have shown that the extremely high electron localization can also be achieved in the dissociation of HD+ and HT+ as that in
Reference
| 1 |
|
| 2 |
|
| 3 |
|
| 4 |
|
| 5 |
|
| 6 |
|
| 7 |
|
| 8 |
|
| 9 |
|
| 10 |
|
| 11 |
|
| 12 |
|
| 13 |
|
| 14 |
|
| 15 |
|
| 16 |
|
| 17 |
|
| 18 |
|
| 19 |
|
| 20 |
|
| 21 |
|
| 22 |
|
| 23 |
|
| 24 |
|
| 25 |
|
| 26 |
|
| 27 |
|
| 28 |
|
| 29 |
|
| 30 |
|
| 31 |
|
| 32 |
|
| 33 |
|