3.1. Simulations of DNA decondensationTypical conformations of DNA chains at varying salt concentration for monovalent, divalent, and trivalent cations are shown in Fig. 1, and three columns display the DNA decondensation for various salt cations. The first column exhibits the changes of DNA/cationic chain complexes with increasing the concentrations of monovalent cations. In the dilute solution, the salt cation concentrations C+ are below 100 mM, such as 13 mM (Fig. 1(a1)) and 63 mM (Fig. 1(b1)), DNA chains keep the condensation state as the attractive interactions between DNA and cationic chains is strong enough in the complexes, and only a few discrete cationic chains are observed. When C+ increases to C+ = 376 mM (Fig. 1(c1)), the added monovalent cations can weaken the attractive interactions between DNA chains and short cationic chains, which can bring about the conformational changes of the complexes. The condensed DNA becomes loose and more short cationic chains get free of the condensed DNA, which can be regarded as the beginning of the DNA decondensation for monovalent salt cations. Furthermore, the added cations can promote the decondensation of DNA chains, as shown in Fig. 1(d1) of C+ = 627 mM, resulting in more discrete cationic chains. At high salt concentrations, like 1253 mM (Fig. 1(e1)), the electrostatic interactions between the added cations and DNA chains can no longer be ignored. Obviously, the DNA chains stretch and short cationic chains are scattered in the simulation box. For the second column, namely the case of divalent salt cations, the conformations of DNA/cationic chain complexes are almost the same as that of monovalent salt cations at low salt concentrations, such as 13 mM (Fig. 1(a2)) and 63 mM (Fig. 1(b2)). With increasing the salt concentration, the intermediate states of the DNA decondensation process can be clearly observed at the concentrations of C+ = 251 mM (Fig. 1(c2)) and 377 mM (Fig. 1(d2)), and at a high salt concentration of C+ = 627 mM (Fig. 1(e2)), the distributions of short cationic chains and DNA chains become uniform. Compared with the results for monovalent and divalent salt cations, trivalent salt cations greatly accelerate the process of DNA decondensation. The DNA chains are also aggregated at low concentrations with C+ < 125 mM (Fig. 1(c3)). The DNA structure looks loose, and the DNA chains are decondensed partially and completely at the concentrations of C+ = 251 mM (Fig. 1(d3)) and 377 mM (Fig. 1(e3)), respectively. Each DNA chain is shown clearly, and the distributions of short cationic chains and DNA chains are uniform. The DNA chains are decondensed at high salt concentrations, and the valence of salt cations affects the process of DNA decondensation. The higher the valence of salt cation is, the easier the compact DNA chains are decondensed.
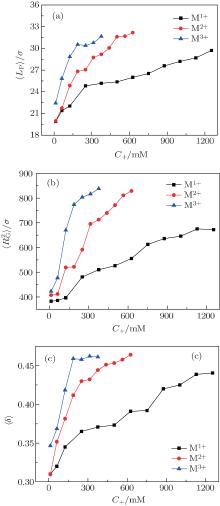
In order to investigate the decondensation processes of DNA chains in more detail, we calculate the average persistence length of DNA chains 〈 LP〉 , the mean-square radius of gyration of DNA chains 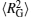 , and the shape factor of DNA chains 〈 δ 〉 to characterize the statistical properties of DNA chains. In Fig. 2(a), we plot 〈 LP〉 as a function of salt cation concentrations C+ for various multivalent cations. The persistence length is employed to characterize the polymers. It is defined as the average sum of the projections of all bonds j ≥ i on bond i in an infinitely long chain.[49] It is given by
, and the shape factor of DNA chains 〈 δ 〉 to characterize the statistical properties of DNA chains. In Fig. 2(a), we plot 〈 LP〉 as a function of salt cation concentrations C+ for various multivalent cations. The persistence length is employed to characterize the polymers. It is defined as the average sum of the projections of all bonds j ≥ i on bond i in an infinitely long chain.[49] It is given by
 | |
where angle θ i is between a vector that is tangent to the polymer at position i and position i − 1, and d represents the length of each monomer of DNA chains. The persistence length of DNA chains is always used to characterize bending stiffness of DNA chains. Here, the values of 〈 LP〉 can also be used to describe the degree of DNA condensation, and three profiles all increase monotonically with increasing salt cation concentrations C+ . However, the slopes of these lines are different and the slope is the largest one for trivalent cations at low concentrations. The fact that the value of 〈 LP〉 increases sharply at low cation concentrations of C+ < 152 mM for trivalent cations shows that the added trivalent cations can accelerate greatly the speed of decondensation for DNA chains at low salt concentrations. Meanwhile, the profile increases slowly at high salt concentrations C+ > 276 mM, which indicates that DNA chains get close to the decondensation equilibrium states. The profiles of divalent salt cations share a similar trend to monovalent ones. The decondensation characteristics of DNA chains can also be confirmed by the mean-square radius of gyration of DNA  , as shown in Fig. 2(b). The values of
, as shown in Fig. 2(b). The values of 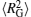 , for divalent and trivalent cations increase sharply at low salt concentrations and slowly at high salt concentrations. However, the value of
, for divalent and trivalent cations increase sharply at low salt concentrations and slowly at high salt concentrations. However, the value of  , for monovalent salt cations increases smoothly with increasing salt cation concentrations. We also introduce shape factor 〈 δ 〉 [50, 51] to describe the structure of condensed DNA, followed by
, for monovalent salt cations increases smoothly with increasing salt cation concentrations. We also introduce shape factor 〈 δ 〉 [50, 51] to describe the structure of condensed DNA, followed by
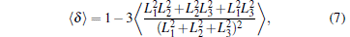 | |
where 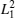 ,
,  , and
, and  are three eigenvalues of diagonal matrix of radius of gyration tensor. The shape factor varies from 0 (sphere) to 1 (rod). Therefore, three monotonic increases in the shape factor of DNA 〈 δ 〉 in Fig. 2(c) can also prove that the more the multivalent salt cations are in the solution, the looser the structure of condensed DNA chains is. Moreover, at the same salt concentration region, the higher the valence of salt cation is, the faster the speed of DNA decondensation is.
are three eigenvalues of diagonal matrix of radius of gyration tensor. The shape factor varies from 0 (sphere) to 1 (rod). Therefore, three monotonic increases in the shape factor of DNA 〈 δ 〉 in Fig. 2(c) can also prove that the more the multivalent salt cations are in the solution, the looser the structure of condensed DNA chains is. Moreover, at the same salt concentration region, the higher the valence of salt cation is, the faster the speed of DNA decondensation is.
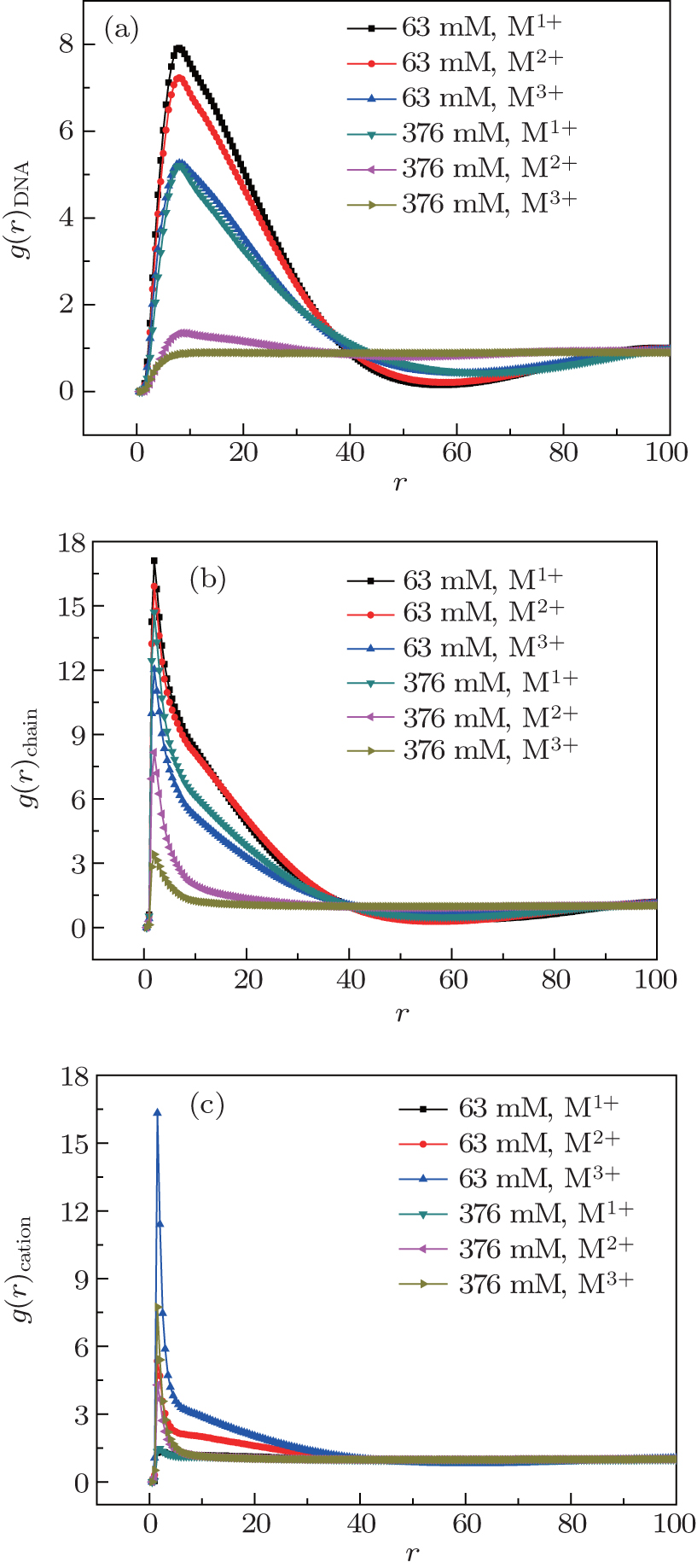
In order to explore the correlation positions between DNA chains and other charged monomers, we calculate the pair correlation functions of DNA– DNA pairs g(r)DNA, DNA– chain (cationic chain) pairs g(r)chain, and DNA– cation pairs g(r)cation for various salt cations, and the results for two cation concentrations of 63 mM and 376 mM are shown in Figs. 3(a)– 3(b), respectively. In Fig. 3(a), we plot the pair correlation function g(r)DNA of the pairs of one DNA monomer and another monomer from other DNA chains as a function of the distance between these two monomers, which can display directly the degree of DNA decondensation. At low cation concentration, there exists a peak for each curve. These peaks are located at r = 8σ , which is close to the length of short cationic chains, and the heights of the peaks are related to the degree of DNA decondensation. As all the DNA chains are charged negatively, there exists the strong repulsive interactions between these DNA chains, and DNA chains do not keep close to each other. DNA chains are aggregated only through adding short cationic chains to the solutions, and the minimum distance between two DNA chains is close to the length of short cationic chains, therefore, all the peaks are located at r = 8σ . The black curve with the highest peak implies that the DNA chains are condensed and the conformations are compact at a concentration of 63 mM for monovalent cations. However, at high cation concentration such as 376 mM, the peaks in the curves disappear completely for trivalent cations and the distribution of DNA chains for trivalent cations is uniform and the decondensation of DNA chains occurs. The pair correlation functions of DNA– chain (cationic chain) pairs g(r)chain can describe directly the distribution of short cationic chains around the DNA chains, and the results are shown in Fig. 3(b). The fact that the sharp peaks are all located at r = 1σ in Fig. 3(b) at low salt concentrations suggests that short cationic chains are connected directly with DNA chains due to the strong attractive electrostatic interactions. At high salt concentrations, the peaks disappear gradually and short cationic chains are away from DNA chains, especially for trivalent cations. As the attractive electrostatic interactions between cationic short chains and DNA chains are weak enough at high salt concentrations, the DNA decondensations take place, especially for trivalent cations. Meanwhile, the peaks in Fig. 3(c) suggest that salt cations are aggregated around DNA chains for high valence cations. When the valence of salt cations increases, the heights of the peaks increase, which is contrary to the results in Figs. 3(a) and 3(b), and this means that the number of short cationic chains around DNA chains increases accordingly. In the meantime, the heights of the peaks decrease when the cation concentrations increase, which are in agreement with the results of salt cations around the dendrimers.[52] In conclusion, the pair correlation functions of DNA– DNA pairs and DNA– cationic pairs confirm directly that cation salt concentrations and cation valence affect the decondensation structures of DNA chains.
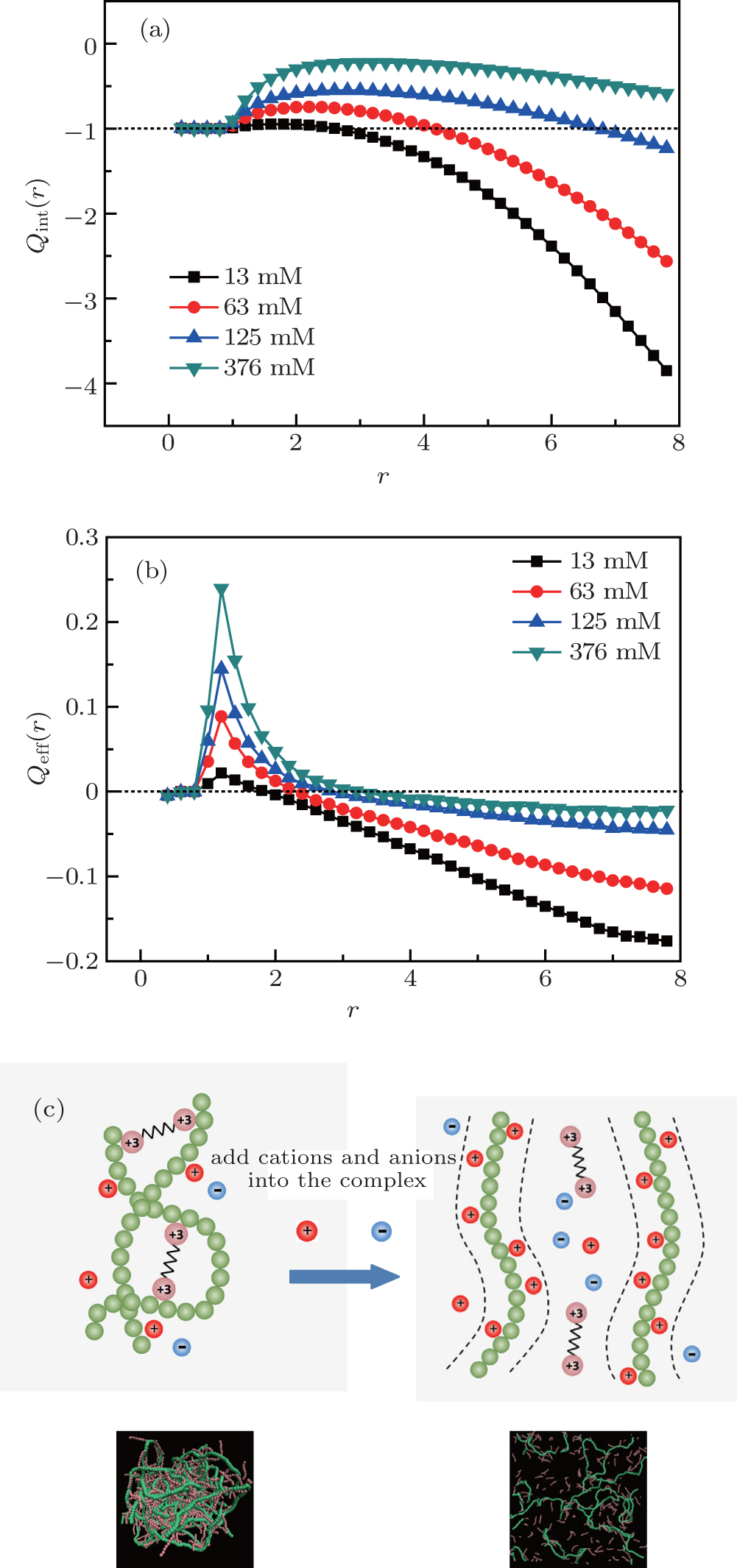
Researchers have attributed the decondensation mechanism to ionic overcompensation[53, 54] or anion incomplete dissociation, [55] in the previous similar studies of decondensation behavior for DNA, [53] single-stranded nucleic acid[54] and single-stranded DNA.[55] Inspired by the three regimes of DNA concentration on monovalent salt concentration proposed by Raspaud el al., [56] we focused on the competition between the cationic chain and added salt ions. Therefore, in order to investigate the reason why the decondensation of DNA chains can occur at high salt concentrations, we calculate the effective charge distributions around DNA chains. We define a wormlike tube around a DNA chain, consisting of N tubes of radius r centered at the bond, and monitor the integrated charge distribution Qint(r) within this tube at various cation concentrations for trivalent cations. The results are shown in Fig. 4(a), and the total charges include the DNA chains, multivalent cations, and anions (counter-ions). The maximum values of Qint(r) increase with increasing the salt cation concentration. At a high salt concentration of C+ = 376 mM for trivalent cations, the integrated charge rises and reaches the maximum at r = 3σ . Although the curve falls down smoothly at this concentration, the values of integrated charges are still larger than − 1.0. This means that there are some net positive charges in the tube around the DNA chains, while at a low salt concentration such as C+ = 13 mM for trivalent cations, the integrated charge Qint(r) rises a little and achieves the maximum at r = 2σ . Then Qint(r) decreases quickly for r > 3.0 and is close to − 4.0 at r = 8.0, which means that there are more counter-ions (anions) in the tube around the DNA chain. In fact, the charges of short cationic chains are not included in calculating the integrated charge distribution Qint(r) because our aim is to calculate the attraction interactions between all the charges near the DNA chains and the short cationic chains. A large negative value of Qint(r) means a strong attractive interaction between short cationic chains and DNA chains because the charges of short cationic chains are not included in calculating the value of Qint(r). As plotted in Fig. 4(b), the local charge density distribution profiles Qeff(r) at various cation concentrations for trivalent cations can represent the charges on a shell whose radius is from r to r + dr (here, dr = 0.2). Obviously, some local regions around DNA are positively charged within 1σ < r < 3σ at high cation concentrations and within 1σ < r < 2σ at low cation concentrations. The height of peaks at high concentration is higher than that at low concentration. Figure 4(c) gives an illustration about the changes of neighboring charges around DNA induced by cations when multivalent cations are added into the complexes. Two insets below are the corresponding simulation snapshots at a low salt concentration (C+ = 13 mM) and a high salt concentration (C+ = 376 mM). When a few cations and anions are added into the complex, the net charges near to DNA chains are negative, see the curve for 13 mM in Fig. 4(b), there exists strong attractive interactions between short cationic chains and DNA chains, and the DNA chains are aggregated through short cationic chains, see the left figure of Fig. 4(c). When the salt concentrations increase, more cations and anions are added into the complexes, some short cationic chains are replaced by some cations. Some cations are located near to DNA chains and some local regions around DNA are positively charged, the attractive interactions between short cationic chains and DNA chains decreases deeply and some short cationic chains are away from the DNA chains. The decondensations of DNA chains take place at high salt concentrations, especially for trivalent cations. The simulation snapshots also show the process of DNA decondensation clearly. It is explicitly demonstrated that cations can overcompensate the bare charge of the DNA chains and weaken the attraction interactions between the DNA chains and short cationic chains at high salt concentrations.
3.2. DNA decondensation in NaCl/MgCl2 solutions The typical AFM images of DNA– spermidine complexes in different solutions with varying salt concentrations are shown in Fig. 5. The top row refers to the NaCl solution, and the bottom one is in the MgCl2 solution, and the salt concentration increases from 0 to 1 M. In the salt free solution, DNA chains are folded and condensed, as shown in Figs. 5(a1) and 5(b1). A high-resolution inset in Fig. 5(a1) captured from the salt free solution exhibits some details of the condensed structure. The bright cores are circled by layers of DNA segments. Unlike the typical toroidal condensates observed in extremely dilute solution, [57] these disk-like or flower-like condensates are observed, especially in the high-resolution inset, which agree well with the results of Ye et al.[58] and Wang et al.[59] As the luminance of AFM images can represent the height of DNA condensates, the white spots in the images are the precipitations of condensed DNA. In the NaCl solution, the white spots are broken a little into smaller ones and the condensed DNA chains are still kept as a whole when the salt concentration increases to 10− 2 M, as shown in Fig. 5(a2). However, when the concentration increases further by one order of magnitude, the condensed DNA chains are broken into some small clusters as shown in Fig. 5(a3), and these clusters go on dividing into smaller ones and parts of the single DNA chain could be observed from the AFM images (see Fig. 5(a4)) when the NaCl concentrations keep increasing. In Fig. 5(a5), in which the salt concentration increases to 1 M/L, we can observe that most of the DNA chains are distinguished with few extremely small white spots and the DNA chains are decondensated completely. The changes of the second row follow the trend of the first one. Compared with the morphologies of DNA chains in NaCl solution, the decondensation behaviors of DNA chains in MgCl2 solution also occur at high concentration. However, it is an obvious difference that when the MgCl2 concentration increases to 1 M, the DNA chains are distributed more homogeneously and each DNA chain is observed more clearly by comparison with the DNA chains at the same concentration in NaCl solution, as shown in Fig. 5(b5). A similar transition of single DNA molecules was reported by Andreia et al., [26] in which an isolated DNA chain was observed, and from the AFM images obtained by other studies, we found DNA chains extending through the mica. However, they achieved that by altering the size of cations[60] or increasing the Zn2+ concentration.[61] In order to know the conformational differences of DNA chains between two solutions at the same concentration, we monitor the mean diameter of DNA condensations at varying salt concentrations in NaCl and MgCl2 solutions, as shown in Fig. 5(b), which can be roughly measured through the plotting scale and the spots in AFM images. The mean diameter of the condensed DNA is related to the degree of DNA decondensation. Two curves start at the same point and decrease with increasing the salt concentration. Compared with the profile of 〈 D〉 in NaCl solution, the curve of 〈 D〉 in MgCl2 solution falls fast at low concentrations. At high concentrations, two profiles tend to be consistent. It is obvious that the average diameter of the DNA condensation in NaCl solution is always larger than that in MgCl2 solution, and the DNA decondensations occur more easily in MgCl2 solution than in NaCl solution, which qualitatively agrees well with our simulation. In a word, the decondensations of DNA chains in DNA– spermidine complexes can be induced by multivalent cations such as Na+ or Mg2+ , and the degree of DNA decondensations depends on the salt cation concentration as well as the valence of cations. The higher the concentration is, the more obvious the DNA decondensation is, and the higher the valence of salt multivalent cations is, the easier the DNA decondensation occurs.






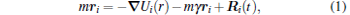

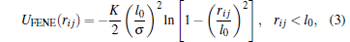

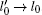

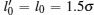
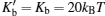





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 , Zhang Lin-Xi
, Zhang Lin-Xi