







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Unconventional phase transition of phase-change-memory materials for optical data storage
Cite this Article
Chen Nian-Ke, Li Xian-Bin. Unconventional phase transition of phase-change-memory materials for optical data storage. Chinese Physics B, 2019, 28(10): 104202 
Permissions
Unconventional phase transition of phase-change-memory materials for optical data storage
† Corresponding author. E-mail:
Abstract
Recent years, optically controlled phase-change memory draws intensive attention owing to some advanced applications including integrated all-optical nonvolatile memory, in-memory computing, and neuromorphic computing. The light-induced phase transition is the key for this technology. Traditional understanding on the role of light is the heating effect. Generally, the RESET operation of phase-change memory is believed to be a melt-quenching-amorphization process. However, some recent experimental and theoretical investigations have revealed that ultrafast laser can manipulate the structures of phase-change materials by non-thermal effects and induces unconventional phase transitions including solid-to-solid amorphization and order-to-order phase transitions. Compared with the conventional thermal amorphization, these transitions have potential superiors such as faster speed, better endurance, and low power consumption. This article summarizes some recent progress of experimental observations and theoretical analyses on these unconventional phase transitions. The discussions mainly focus on the physical mechanism at atomic scale to provide guidance to control the phase transitions for optical storage. Outlook on some possible applications of the non-thermal phase transition is also presented to develop new types of devices.
Keyword:light-matter interaction;phase-change memory;non-thermal phase transition;optical data storage
1. Introduction
Phase-change memory (PCM) was proposed by Stanford Ovshinsky in 1968.[1] The information storage of PCM relies on the big contrast of the electronic or optical properties between the crystalline phase (with low resistance and high reflectance) and the amorphous phase (with high resistance and low reflectance) of PCM materials. The data writing and erasing operations are realized by the reversible phase transitions between the two phases. Since the operating stimuli also can be electronic or optical pulses, it can be used for both electronic and optical memory technologies.[2–5] PCM is firstly commercialized as optical memory including CD, DVD, and BD-RE.[6] These optical disks have been the main media for data storage for about 20 years. During the last two decades, the electronic devices, including computers, mobile phones, and so on, developed much faster than optical devices. Accordingly, the electronic PCM devices gradually became the main stream of PCM technology. Recently, however, the optically controlled phase-change memory restores the vitality due to the emergence of new applications, such as integrated all-optical non-volatile memory,[7] optically controlled in-memory computing,[8] and brain-inspired computing,[9,10] (see Fig.
 | Fig. 1. The applications of phase-change memory in optical memory and in-memory/neuromorphic computing. (a) The integrated all-optical non-volatile memory based on PCM material Ge2Sb2Te5 (GST). Here, the PCM material is placed on the top of a waveguide. The data recording is realized by the optical near-field effects. The writing and erasing operations are achieved by the laser pulse passes through the waveguide while the read operation relies on the transmission signal modulated by the optical absorption of PCM materials. (b) The illustration of in-memory arithmetic computing by optically-controlled phase change. (c) The all-optical neuromorphic computing system based on PCM material GST. The input pulses can induce the phase change (recrystallization) of PCM material, which in turn changes the output signals. Both the in-memory computing and the neuromorphic computing utilize the accumulation effect of the crystallization. Panel (a) is reproduced from Ref. [7] with permission from the Nature Publishing Group. Panel (b) is reproduced from Ref. [8] with permission from the John Wiley and Sons. Panel (c) is reproduced from Ref. [9] with permission from the Nature Publishing Group. |
Although the data processing at present is mainly dealt by electronic computers, the data transmission is dominated by optical communications in optical fibers. Therefore, repeated conversions between the electronic and the optical information are needed. If the data can be directly dealt by optical computing technology, the processing efficiency will be significantly enhanced. Moreover, the power consumption of optical computing also is believed to be lower than that of electronic computing.[11] As demonstrated by previous progresses,[7–9] optically controlled PCM should be a promising candidate for the advanced memory or computing technologies in the future. In addition, the laser-induced phase transition of PCM materials also shows potential in some other novel applications, such as the thermal emitter,[12] the thermal camouflage,[13] the plasmonic beam switching of metasurface and the bifocal lensing.[14] New opportunities are often companied by new challenges. Either in-memory computing or brain-inspired computing needs a fine control of the structures, properties and the phase-change dynamics of PCM materials. In this article, we review the understanding of laser-induced phase transitions in PCM materials which is the key for this technology. The dynamic process, the speed, the property change, the stability, the reversibility, and the cyclability of the transition closely influence the performances of PCM devices. Optical PCM involves both thermally and non-thermally driven phase transitions while electronic PCM mainly drives by Joule heating. Owing to the absence of violent thermal melting, the non-thermal phase transitions show some superior performances on the fine control of PCM materials by achieving unconventional phase transitions. However, the underlying mechanisms are still not fully understood due to the complicated light–matter interactions. Here, we mainly focus on the dynamic mechanism of laser-induced non-thermal phase transitions at atomic scale to provide fundamental supporting for optically controlled phase-change memory or computing.
2. Solid-to-solid amorphization induced by optical excitation
For optical PCM, the stimuli is laser pulse and the signal contrast (0/1) comes from the large difference in optical reflectance between the two phases (see Fig.
 | Fig. 2. The traditional mechanism of the melt-quenching and the recrystallization processes. The crystalline phase possesses a high reflectance while the amorphous phase possesses a low reflectance. |
However, in 2011, by first-principles calculations, Li et al. proposed that a non-thermal effect of the optical excitation played important roles in the ultrafast laser-induced amorphization in PCM materials.[25,26] When electrons are excited from valence band to conduction band, the chemical bonds are weakened due to the electron transfer from bonding states to anti-bonding states. Such an instability mechanism also has been suggested in the femtosecond (fs) laser-induced disordering of Si and GaAs.[27,28] The results of the ab initio molecular dynamics (AIMD) simulation in Figs.
 | Fig. 3. Electronic-excitation-induced solid-to-solid amorphization in GST. (a)–(f) Structural snapshots during the AIMD simulation of amorphization under a 9% excitation. The crystalline structure turns to a disordered phase within 24 picoseconds (ps). (g) Mean square distances (MSD) during the AIMD simulations of the excitation-induced amorphization and the normal melting. The blue line indicates the exited state (Exc.) while the red line represents the melting state (Mel.). (h) The change of amorphization temperatures with different excitation intensities. The data are obtained from a series of AIMD simulations with different excitation intensities and temperatures. The red line is a quadratic fitting. (i) Partial density of states of GST. Upon a laser excitation, electrons are mainly excited from valence band maximum, i.e., the region labelled by “j”. Panels (j) and (k) show the projected electronic density distribution of the energy regions indicated by “j” and “k” in panel (i), respectively. Obviously, for the band-edge excitation, Ge atoms contribute more electronic states than Sb atoms. Panels (a)–(g) and (i)–(k) are reproduced from Ref. [26] with permission from the American Physical Society. Panel (h) is reproduced from Ref. [25] with permission from the John Wiley and Sons. |
The non-thermal amorphization is also supported by some recent experiments. For example, Konishi et al. have reported that the fs laser irradiation in Ge10Sb2Te13 can result in an ultrafast (∼500 fs) decrease of the reflectivity [see Fig.
 | Fig. 4. Femtosecond (fs) laser-induced amorphization of PCM materials. (a) Time-resolved reflectivity of Ge10Sb2Te13 after a 140-fs laser irradiation. The reflectivity intensity of PCM material decreases within 500 fs and then maintains for at least several tens of ps. (b) Scanning electron micrograph of a GST surface after irradiated by an 18.1-mJ/cm2 fs pulse. The dotted circle indicates the irradiated area where grain boundaries can be clearly seen. (c) Time-resolved reflectivity of the GST upon an fs laser irradiation. The data of different fluences are adopted from different areas of a light spot. The red dashed line indicates the reflectivity of liquid phase. (d) Time-resolved white line intensities of XANES of GST during the 600-ps laser-induced amorphization (red dots and line). Obviously, the intensities during amorphization are still far from the intensity of liquid state. Panels (a) and (b) are reprinted with permission from Ref. [29] ©The Optical Society. Panel (c) is reproduced from Ref. [30] with permission from the AIP publishing. Panel (d) is reproduced from Ref. [33] with permission from the American Physical Society. |
Since optical reflectivity only indicates an electronic structure,[32] Fons et al. have used the white line intensity of x-ray absorption near edge structure (XANES) to detect the amorphization process via structural information.[33] Obviously, the results in Fig.
Recent years, time-resolved x-ray/electron diffractions have been utilized to directly catch the dynamic structural transitions. Waldecker et al. compared the time-resolved signals of dielectric function and x-ray diffraction (XRD) of GST under an fs laser irradiation [see Figs.
 | Fig. 5. Time-resolved (a) imaginary part of the dielectric function and (b) XRD intensity of GST after the fs laser irradiation. The intensity of the dielectric function dramatically decreases and then recovers to a certain degree which seems to be a behavior of electrons. In contrast, the intensity of XRD decreases slowly but remains stable which is regarded as the change of structures. (c) Schematic illustrations of the excitation-induced intermediate phase by structural rotation. Panels (a) and (b) are reproduced from Ref. [35] with permission from the Nature Publishing Group. Panel (c) is reproduced from Ref. [36] with permission from the Nature Publishing Group under a Creative Commons Attribution 4.0 International License (CC BY 4.0). |
3. Mechanical effects of optical excitation
Here,
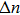
is the excited electron density, and
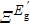
is the effective deformation potential induced by an excitation. In GST, when the excitation intensity is larger than 6%, the effect of the stress is to drive the volume expansion [see Fig. 6(a) ]. According to Eq. (1 ), the stress by excitation should be in proportion to the effective deformation potential and excitation intensity which has been confirmed by the calculations in a series of semiconductors [see Fig. 6(b) ]. Since diamond and BN have very large deformation potentials, their excitation stresses are extremely large. Therefore, the excitation stress should be a general effect that is adapted for other materials.
In addition to the local atomic motions, the optical excitation can also lead to global volume changes. For example, by time-resolved x-ray diffraction, Fons et al. detected a large volume expansion in the fs laser-irradiated Ge2Sb2Te5.[38] Their analyses suggested that the volume expansion cannot be explained solely by a thermal expansion. The first-principles calculations also proposed a giant expansion effect of electronic excitations. The excitation-induced expansion can be explained by the quantum electronic stress effect [see Figs.
 |
 | Fig. 6. (a) The stresses and corresponding volume changes as a function of excitation intensity in rock-salt GST. Here, the negative stress leads to lattice expansions. (b) The stress as a function of the excitation intensity in a series of semiconductors. The inset shows the schematic illustration of inter-band excitation. CB and VB mean the conduction band and valence band, respectively. (c) The projected forces on individual atoms under a 15% excitation in rock-salt GST. The length of arrows indicates the magnitudes. (d) The magnitude of forces on Te atoms with different neighboring vacancies and excitation intensities in rock-salt GST. All panels are reproduced from Ref. [39] by permission of the PCCP Owner Societies. |
To understand the rule of local atomic motions upon excitation, forces on individual atoms were studied by first-principles calculations in crystalline GST.[39] The local forces in rock-salt GST under the excitation display random directions [see Fig.
4. Order-to-order phase transition induced by optical excitation
Although non-thermal phase transition has been demonstrated in some cases, the final result is usually an amorphization that is similar to the case by the thermal melt-quenching process. Surprisingly, in the most recent reports, Hu et al. and Matsubara et al. have observed an fs laser-induced rhombohedral-to-cubic phase change in GeTe by time-resolved electron diffraction and XRD, respectively [see Figs.
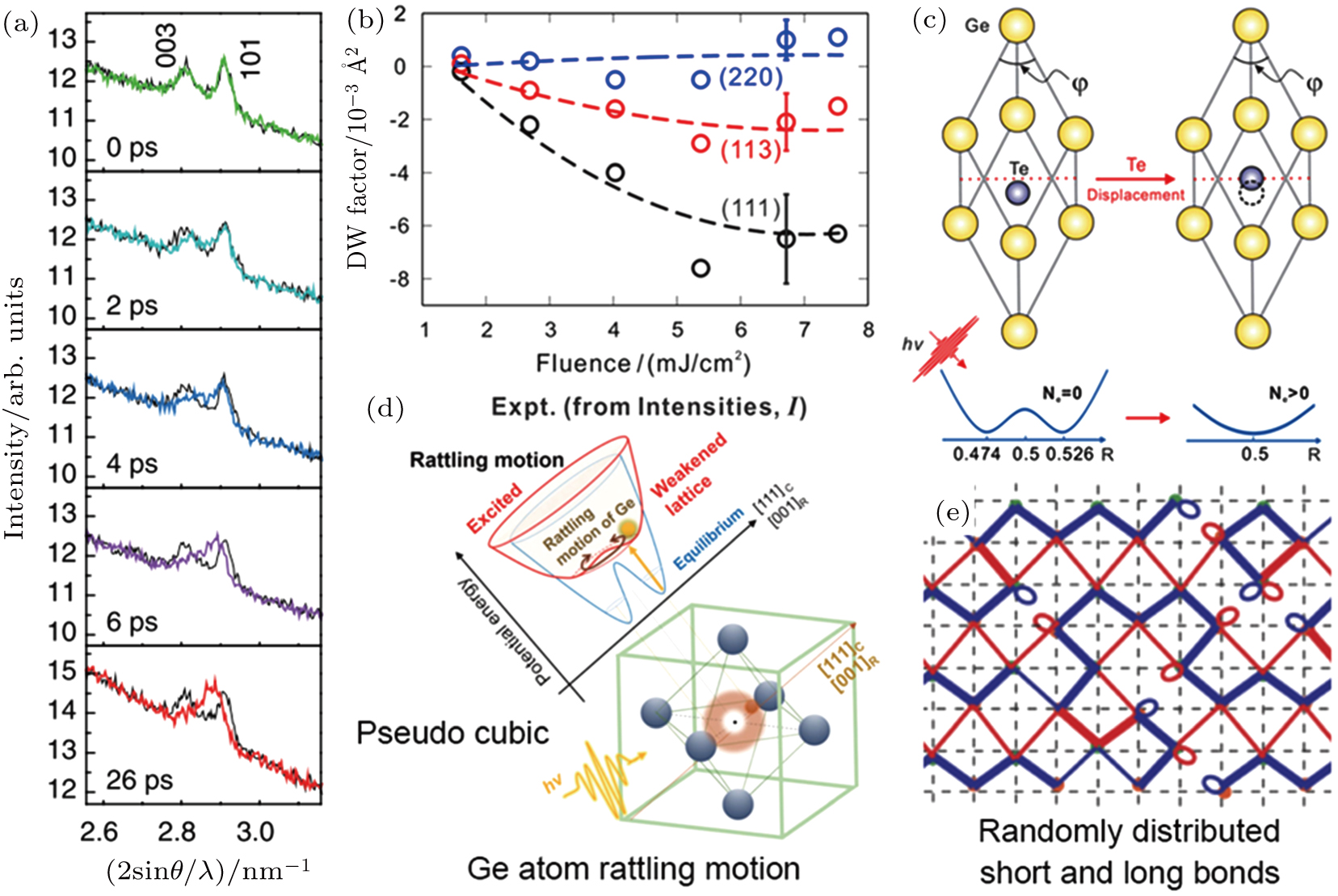 | Fig. 7. (a) Time-resolved XRD spectra of GeTe after fs-laser irradiations. The black lines are the XRD spectra before excitation for comparing. The merging of (003) and (101) peaks indicate the symmetry transition from rhombohedral to cubic. (b) The Debye–Waller factor deduced from the intensity of electron diffraction as a function of laser fluence. By comparing with theoretical simulations, the change of intensities is also demonstrated to be an indication of the symmetry transition from rhombohedral to cubic. (c) The schematic illustration of Te-atom displacement model where the cubic symmetry is a direct result of Te-atom movement. (d) The schematic illustration of Ge-atom rattling model where the Ge atoms dynamically move around the center of cubic sites. The average effect leads to a signal of cubic symmetry. (e) The schematic illustration of randomly distributed long and short bonds. The diffraction signal of such a structure also shows a cubic symmetry. Panels (a) and (d) are reproduced from Ref. [42] with permission from the American Physical Society. Panels (b) and (c) are reproduced from Ref. [41] with permission from the American Chemical Society. Panel (e) is reproduced from Ref. [43] with permission from the American Chemical Society. |
To clarify the debates, Chen et al. performed first-principles calculations and real-time time-dependent density functional theory molecular dynamics (TDDFT-MD) simulations for GeTe by optical excitation.[44] TDDFT-MD is an advanced method to properly describe the excited-state behaviors because it contains the non-adiabatic electron–electron and electron–phonon couplings that beyond the traditional molecular dynamics simulations based on Born–Oppenheimer approximation.[45,46] The simulation with a 5% excitation intensity successfully reproduces the experimentally observed rhombohedral-to-cubic phase transition (Fig. 

 | Fig. 8. (a) The electron density distribution upon the 5% excitation in rhombohedral GeTe. The red arrows indicate the force acting on atoms. (b) The change of potential energy surface of GeTe due to electronic excitation. (c) The trajectories of atomic motions during the TDDFT-MD simulation of the phase transition in GeTe with a 5% excitation. Time evolution of (d) average force, (e) bond length, (f) temperature, (g) kinetic energy, and (h) momentum during the TDDFT-MD in GeTe under a 5% excitation. All panels are reproduced from Ref. [44] with permission from the American Physical Society. |
Moreover, the kinetic energy and momentum of Ge and Te atoms that projected in three dimensions (x, y, z) show a strikingly higher component along [001] axis or z direction due to the excitation [see Figs.
5. Conclusion and perspectives
In this work, some recent progress on ultrafast laser-induced unconventional phase transitions in phase-change-memory materials are summarized to the community. Traditional understanding on the laser-induced amorphization is a thermally driven melt-quenching process. As the laser pulse duration becomes very short (e.g., femtosecond scale), the non-thermal effect of electronic excitation would play significant roles in the phase changes. First-principles calculations reveal the solid-to-solid amorphization in GST under optically electronic excitation. Analysis on electronic structures suggests that electronic excitation drives Ge atoms to move from the octahedral to the tetrahedral sites and thus triggers the amorphization very fast. Non-thermal amorphization of GST without melting is also supported by a series of experiments. Time-resolved reflectivity obtained by the pump-probe technology shows an amorphization process of GST without experiencing a liquid phase. Time-resolved XRD also shows locally structural changes of GST without melting, including local distortions and volume expansions. Furthermore, two recent experiments with time-resolved XRD and electron diffraction techniques both observed an order-to-order (rhombohedral-to-cubic) phase transition in GeTe by fs laser which is in contrast to the usual amorphization of PCM materials induced by laser. Due to the absence of atomic pictures in real time and real space, the underlying mechanism has been intensely debated. By TDDFT-MD, the atomic driving force and phase change dynamics are revealed. Owing to the change of potential energy surface upon electronic excitation, directional forces along [001] axis are instantly produced which directly drives the phase transition. The molecular dynamics simulations suggest that the order-to-order phase transition in GeTe can happen within 1 ps because it is completed by coherent atomic motions without the nucleation-and-growth process and the atomic diffusions. These unconventional phase transitions in PCM materials show the ability of ultrafast laser to manipulate structure beyond thermal melting. Compared with the conventional amorphization via a melt-quenching process, the solid-to-solid amorphization involves much less atomic diffusions. Moreover, the order-to-order phase transition not only possesses a speed with about 3 orders of magnitude faster than conventional phase-change random access memory, but also can control the property of phase-change materials with minimal atomic movements. Therefore, these unconventional phase transitions could be a promising route to promote optical storage and future optical computing with faster speed, lower power consumption, and better endurance.
Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] | |
| [32] | |
| [33] | |
| [34] | |
| [35] | |
| [36] | |
| [37] | |
| [38] | |
| [39] | |
| [40] | |
| [41] | |
| [42] | |
| [43] | |
| [44] | |
| [45] | |
| [46] |