
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Tensile-strain induced phonon splitting in diamond
Cite this Article
Pu Meifang, Zhang Feng, Liu Shan, Irifune Tetsuo, Lei Li. Tensile-strain induced phonon splitting in diamond. Chinese Physics B, 2019, 28(5): 053102 
Permissions
Tensile-strain induced phonon splitting in diamond
† Corresponding author. E-mail:
Abstract
Abstract
The first-order Raman spectroscopy of diamond exhibits splitting and redshift after the burst of high-pressure (160–200 GPa) and high-temperature (∼2000 K). The observed longitudinal optical (LO) and the transverse optical (TO) splitting of Raman phonon is related to the tensile-strain induced activation of the forbidden or silent Raman modes that arise in the proximity of the Brillouin zone center.
1. Introduction
Raman scattering can provide important information on lattice vibrational properties of the chosen materials of interest. For intrinsic crystals, only phonon modes in the proximity of the Brillouin zone (BZ) center are observed owing to the phonon wave vector 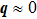
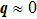
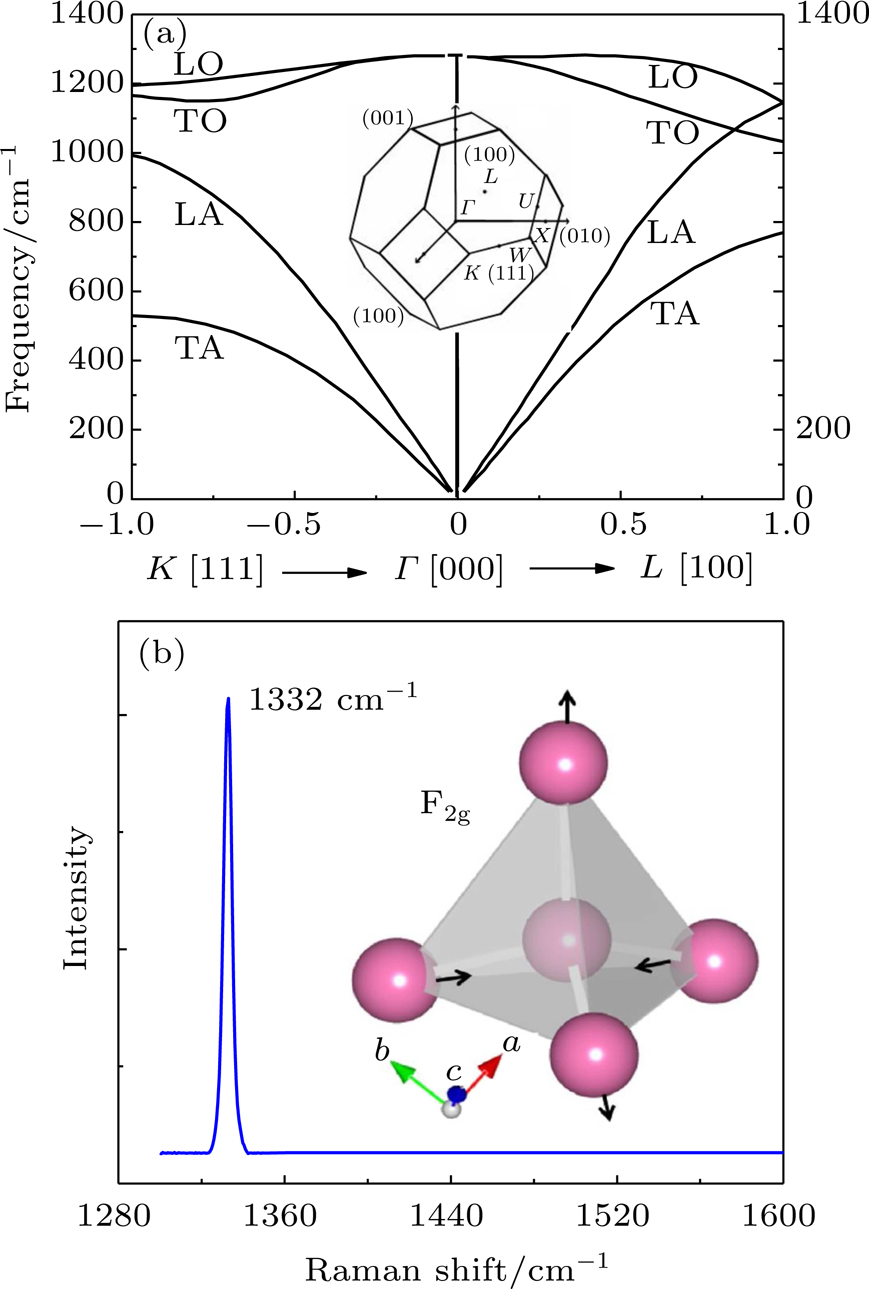
The first-order phonons of diamond can be classified in the space group Fd-3m.[10,11] There are two carbon atoms in the primitive unit cell and thus a single triply degenerate first-order phonon with symmetry 
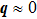

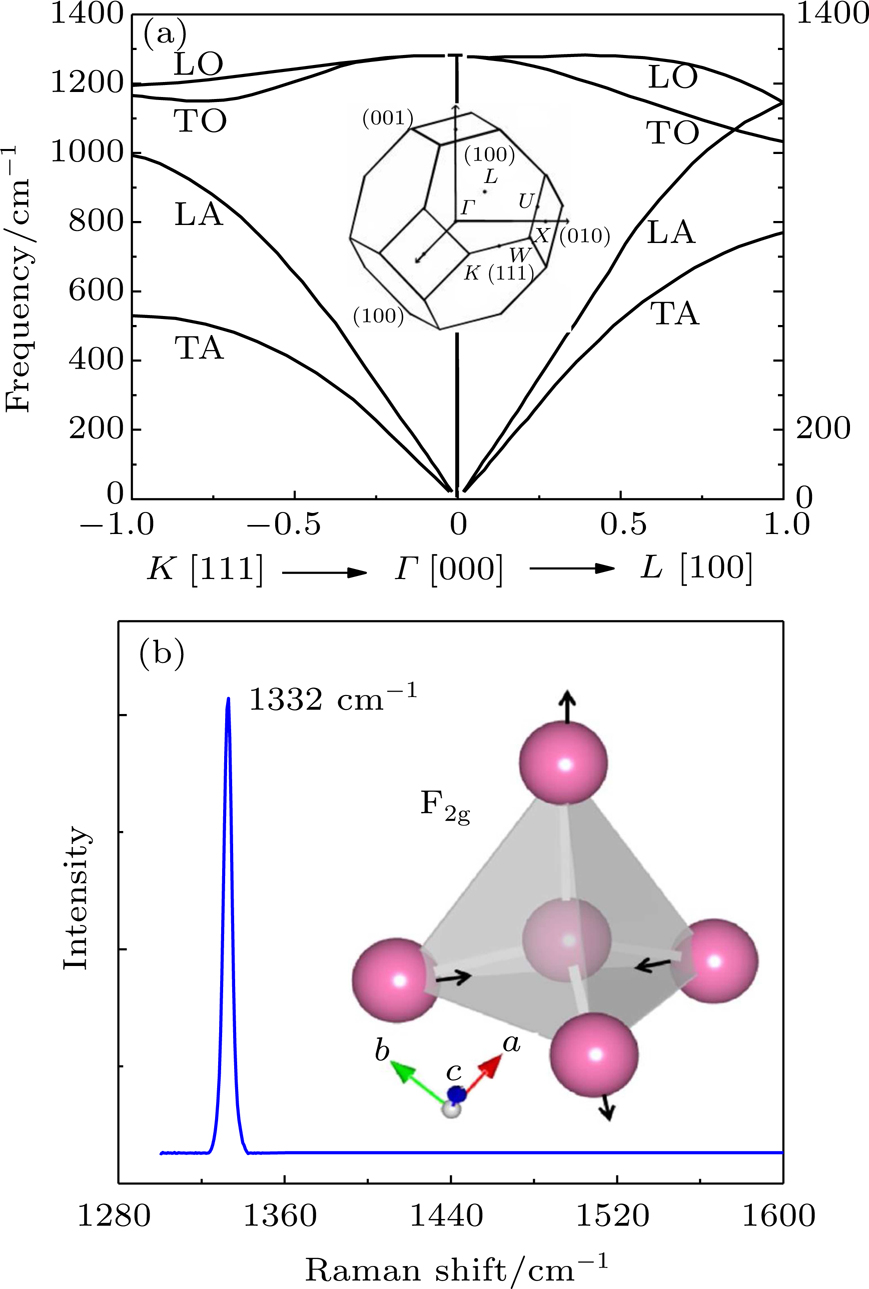 | Fig. 1. (a) Dispersion curves for diamond in the (100) and (111) directions and Brillouin zone of cubic diamond. (b) A first-order fingerprint Raman peak at 1332 cm−1 and the Raman vibrational motions F2g of diamond. |
Stress-induced lattice strain and distortion violates the translational invariance and breaks down the phonon wave-vector selection rule so that the phonon's mode off the center of the BZ can be active in Raman scattering. The question of whether or not the disorder-activated phonon behavior could retain some in situ high-pressure characteristics after the shock treatment has, therefore, to be addressed in order to better understand the dynamic of the aforementioned lattice distortion.
In this paper, we study the corresponding diamond phonon behavior under the shock treatment of high-temperature and high-pressure. We reveal the mechanism of disorder-activated Raman (DAR) phonon originated from the presence of tensile strain in the diamond lattice.
2. Experimental
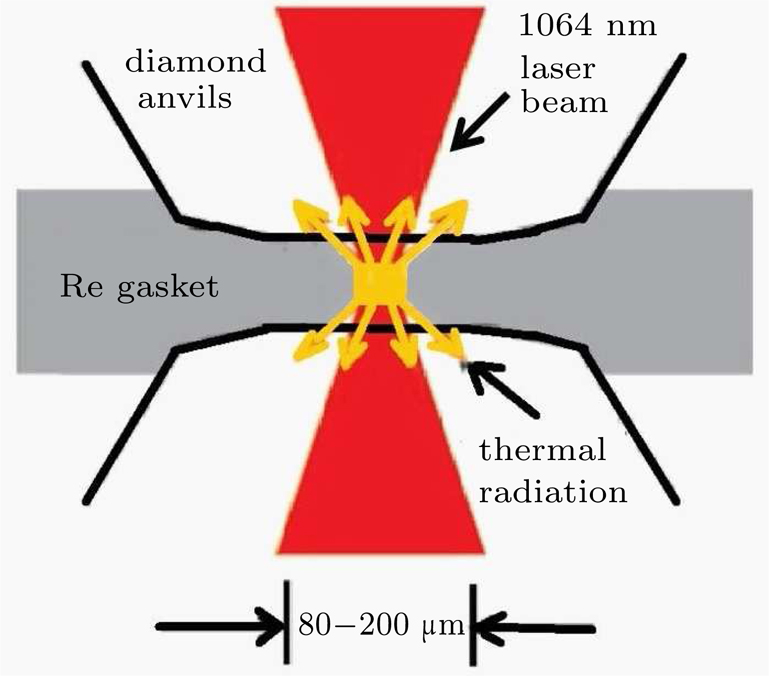
The ultrahigh-pressure Raman scattering experiment and double-sided laser-heated diamond anvil cell (LHDAC) experiment of this work were performed in Sichuan University. The pressure-generation experimental runs using a DAC were performed with a gasketed high-pressure technique.[20] We used the high-frequency edge of the diamond phonon as the pressure scale.[18] In this study, low-luminescence type IA diamonds with a culet diameter of 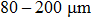
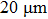


 | Fig. 2. Scheme of the double-side LHDAC experiments. There is a small sample chamber for laser heating, typically smaller than 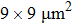
|
Raman scattering experiments were carried out on a custom-built confocal Raman spectrometry system in the backscattering geometry based on triple grating monochromator (Andor Shamrock SR-303i-B, EU) with an attached electron multiplying charge-coupled device (EMCCD, ANDOR Newton DU970P-UVB, EU); excitation was achieved by using a solid-state laser at 532 nm (RGB laser system, NovaPro, Germany) with a laser power of 50 mW for Raman measurement and collection by a 20×, 0.28 numerical aperture (N.A.) objective (Mitutoyo, Japan). The spectral resolution was within ±1 cm−1, and the spatial resolution was within 
3. Results and discussions
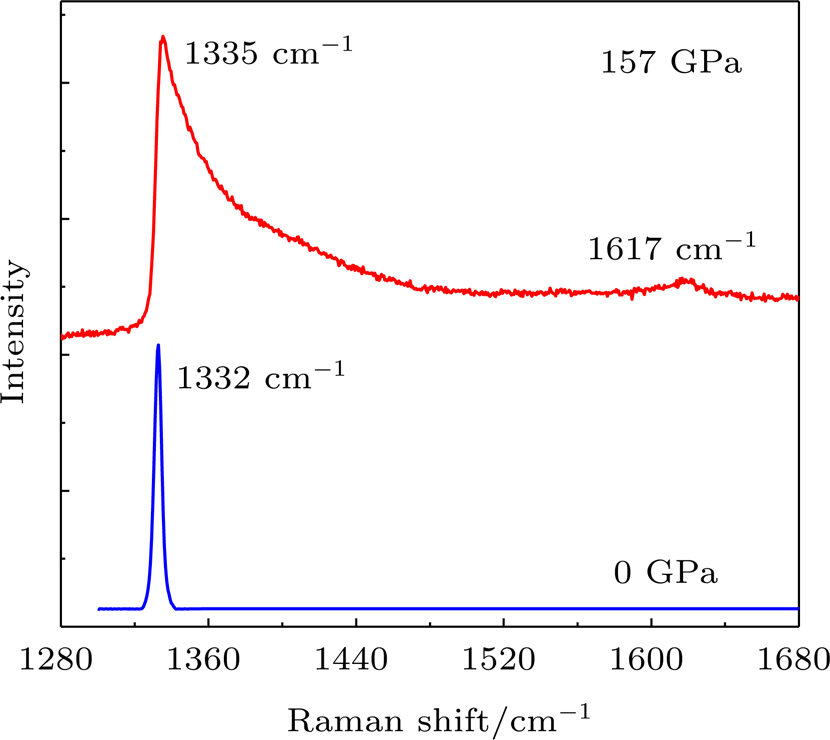
With applied pressure, the Raman shift of the culet center of loaded diamond anvils is found to exhibit asymmetric broadening and blue-shift (due to the normal compressive stress). In marked contrast to the intrinsic Raman line at 1332 cm−1, the Raman band of the diamond anvils loaded at 157 GPa exhibits significant broadening and blue-shift (Fig.
 | Fig. 3. Typical Raman spectra from the center of the diamond anvil culet at 0 GPa and 157 GPa. |
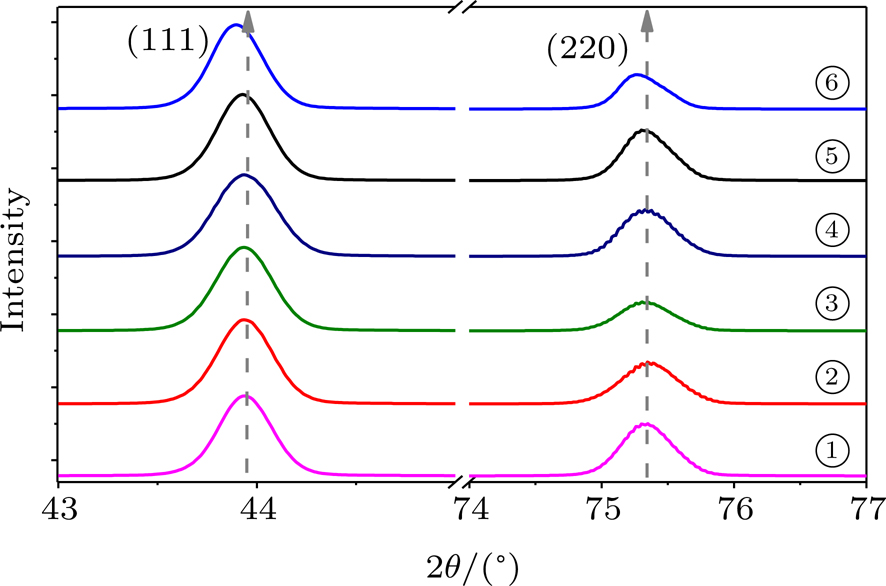
 | Fig. 4. The SEM images of recovered diamond anvils. The white number calibrates the test points of the sample, which corresponds to the Raman and micro-XRD measurements. |
Figure
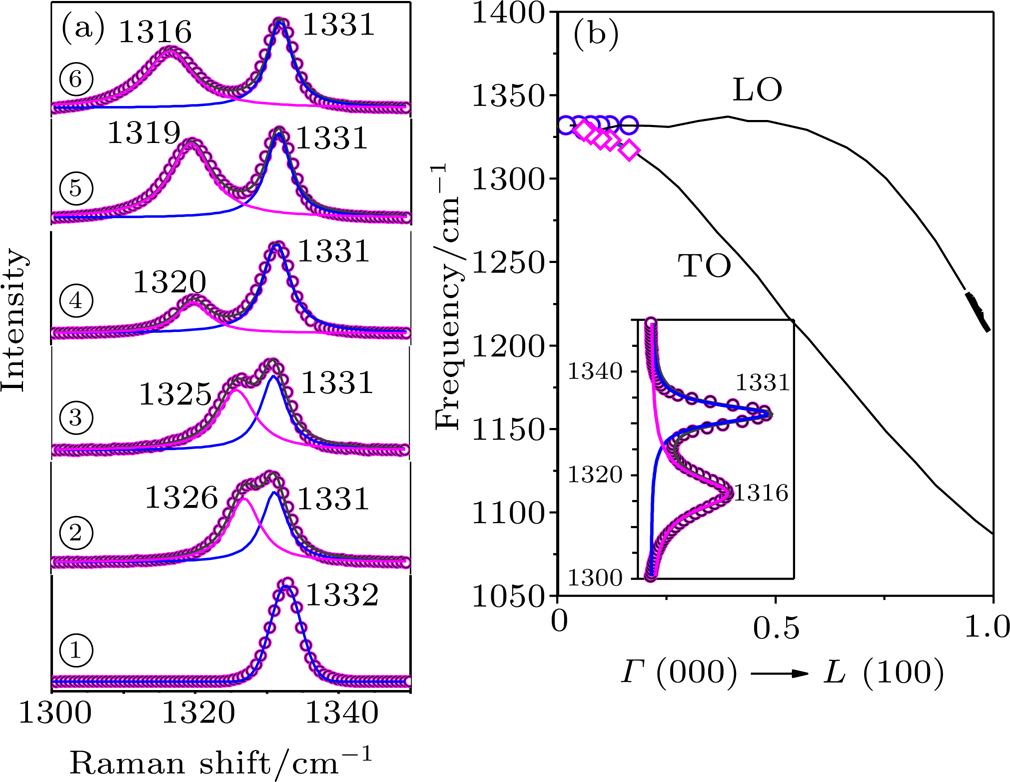
 | Fig. 5. (a) Raman spectra of the recovered diamond anvil for different sample regions and first-order Raman band splitting into a doublet band of 1316–1331 cm−1. It is a consistent one-to-one match between the positions in SEM image (fig. |
Considering that Raman excitation light has a very large wavelength (532 nm) when compared to the distance between neighboring atoms in a diamond crystal (∼0.154 nm), the momentum conservation selection rule basically implies that only phonons at BZ center (
The phonon modes (lattice vibrations) are usually presented by the irreducible representation based on the symmetry group of the crystals, which are essential to the interpretation of Raman spectra. The Raman shift is given by the phonon vibrational frequency, which is governed by the mass of the atoms and the strength of the chemical bonds. In our experiments, the first-order Raman peak splitting induced by tensile strain is consistent with the splitting of transverse optical (TO) and longitudinal optical (LO) in the first Brillouin zone, as shown in Fig. 
Further evidence on tensile strain in the diamond lattice is provided by micro-focused XRD measurements. Figure 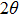
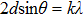
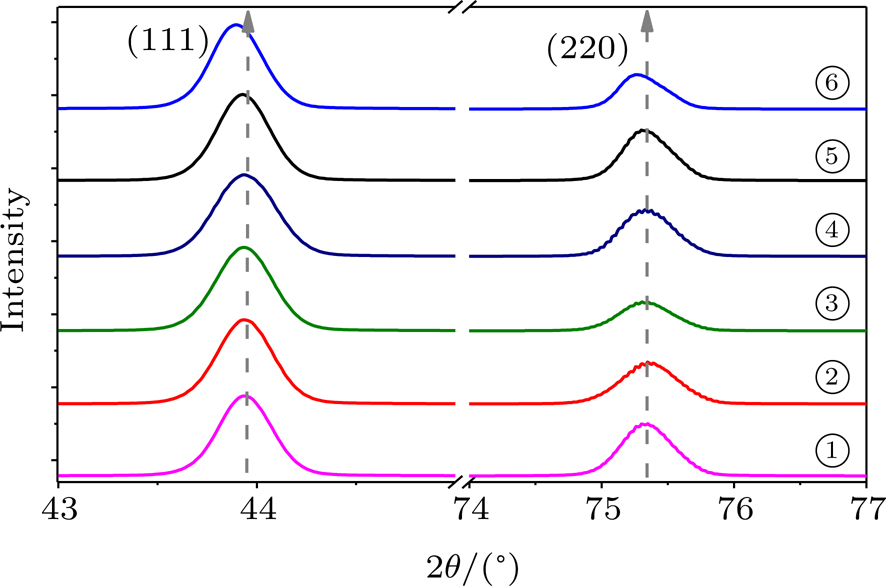 | Fig. 6. Micro-focused XRD of the recovered diamond anvil for different sample regions. It is a consistent one-to-one match between the positions in SEM image (fig. |
4. Conclusions
The phonon splitting behaviors of diamond after a burst of high-pressure and high-temperature have been investigated by micro-Raman spectroscopy, micro-focused XRD, and SEM analysis. The double-splitting peak of diamond is due to the presence of tensile strain in the diamond lattice.
Acknowledgments
We thank Prof. Filippo Boi for helpful discussions and the Joint Usage/Research Center PRIUS (Ehime University, Japan).
Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] |