{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Luminescent properties of thermally activated delayed fluorescence molecule with intramolecular π–π interaction between donor and acceptor
Cite this Article
Cai Lei, Fan Jianzhong, Kong Xiangpeng, Lin Lili, Wang Chuan-Kui. Luminescent properties of thermally activated delayed fluorescence molecule with intramolecular π–π interaction between donor and acceptor
. Chinese Physics B, 2017, 26(11): 118503 
Permissions
Luminescent properties of thermally activated delayed fluorescence molecule with intramolecular π–π interaction between donor and acceptor
† Corresponding author. E-mail:
Project supported by the National Natural Science Foundation of China (Grant Nos. 11374195 and 21403133), Taishan Scholar Project of Shandong Normal University, China, the Promotive Research Fund for Excellent Young and Middle-aged Scientists of Shandong Province, China (Grant No. BS2014cl001), and the China Postdoctoral Science Foundation (Grant No. 2014M560571).
Abstract
Influence of intramolecular π–π interaction on the luminescent properties of thermally activated delayed fluorescence (TADF) molecule (3, 5-bis(3,6-di-tert-butyl-9H-carbazol-9-yl)-phenyl)(pyridin-4-yl) methanone (DTCBPY) is theoretically studied by using the density functional theory (DFT) and time-dependent density functional theory (TD-DFT). Four conformations (named as A, B, C, and D) of the DTCBPY can be found by relax scanning, and the configuration C corresponds to the luminescent molecule detected experimentally. Besides, we calculate the proportion of each conformation by Boltzmann distribution, high configuration ratios (44% and 52%) can be found for C and D. Moreover, C and D are found to exist with an intramolecular π–π interaction between one donor and the acceptor; the intramolecular interaction brings a smaller Huang–Rhys factor and reduced reorganization energy. Our work presents a rational explanation for the experimental results and demonstrates the importance of the intramolecular π–π interaction to the photophysical properties of TADF molecules.
PACS:
85.60.Bt
Keyword:thermally activated delayed fluorescence;intramolecular π-π interaction;Huang-Rhys factor and reorganization energy;aggregation induced enhanced emission
1. Introduction
Great progress has been made in the last three decades on the development of organic light-emitting diodes (OLEDs).[1–4] As the third generation of OLEDs, the thermally activated delayed fluorescence (TADF) materials have received extensive attention in recent years[5–10] since the pioneering work was performed by Adachi and co-workers in 2012.[11–13] TADF materials are pure organic molecules which are cheaper than phosphorescence materials and will not cause environment pollution. Besides, TADF materials with small energy gap (ΔEST) between the lowest singlet excited state (S1) and the lowest triplet excited state (T1) can make full use of excitons and achieve nearly 100% internal quantum efficiency (IQE). With the great progress in TADF-OLEDs, the external quantum efficiency (EQE) of the OLED device has reached up to 30%, which significantly breaks the efficiency limitation of fluorescent OLEDs.[14,15] As TADF molecules are usually formed by donors and acceptors, large systems may have more than one steady configuration.[10,16] When they are prepared in the OLED devices, aggregation-caused quenching (ACQ), aggregation-induced emission (AIE), and aggregation induced enhanced emission (AIEE) phenomena can be observed due to the intermolecular interaction.[17–19] Numbers of studies of the intermolecular interaction on the luminescence properties have been performed,[20] while the intramolecular interaction on the light-emitting properties is seldom studied.[21,22] It is found that the performance of the emitters can be improved by reducing the intramolecular π–π stacking. The intramolecular interaction can be impacted through adjusting the distance between the donors and in turn can change the molecular luminescence properties.[21] How do the π–π interaction between the donors and acceptors influence the luminescence properties? There has been no report until now.
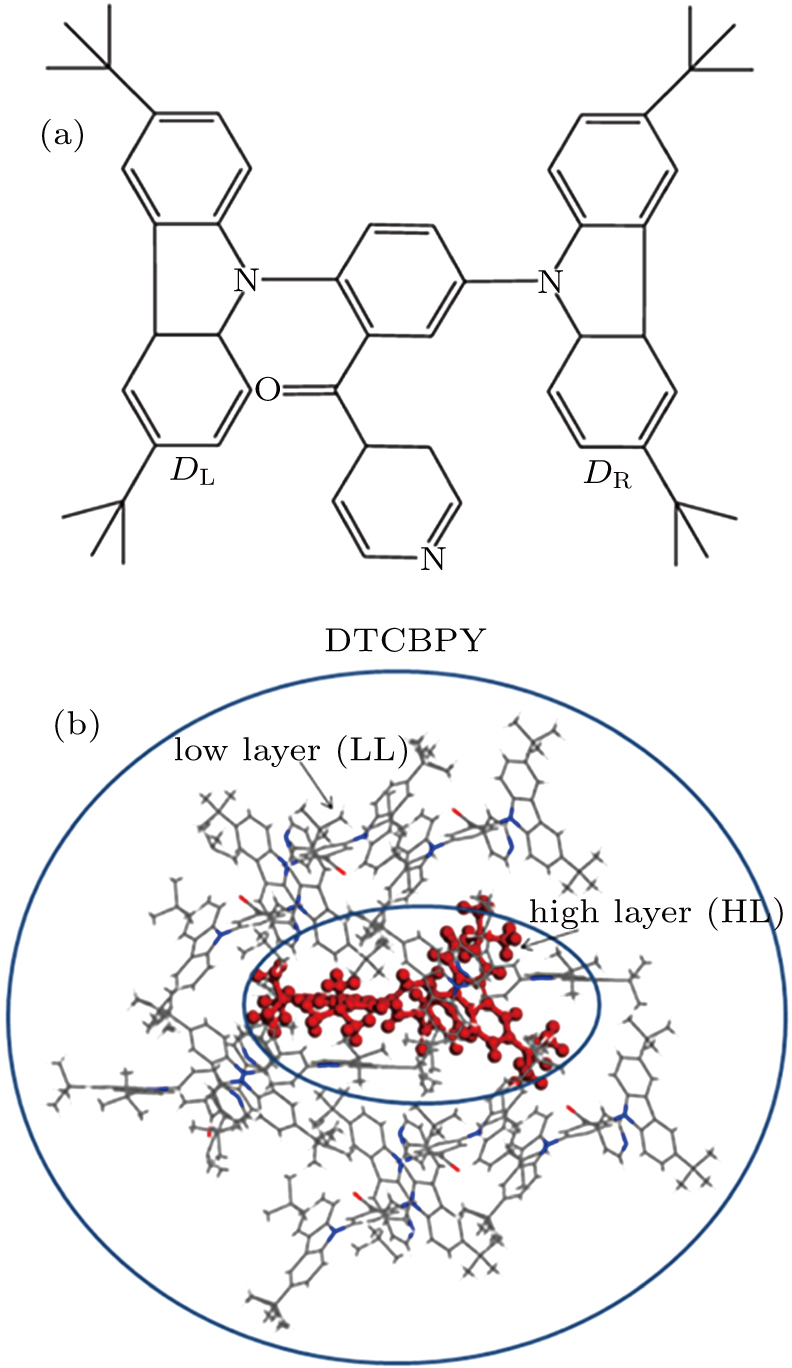
Recently, a molecule named (3,5-bis(3,6-di-tert-butyl-9H-carbazol-9-yl)-phenyl) (pyridin-4-yl) methanone (DTCBPY) (as shown in Fig.
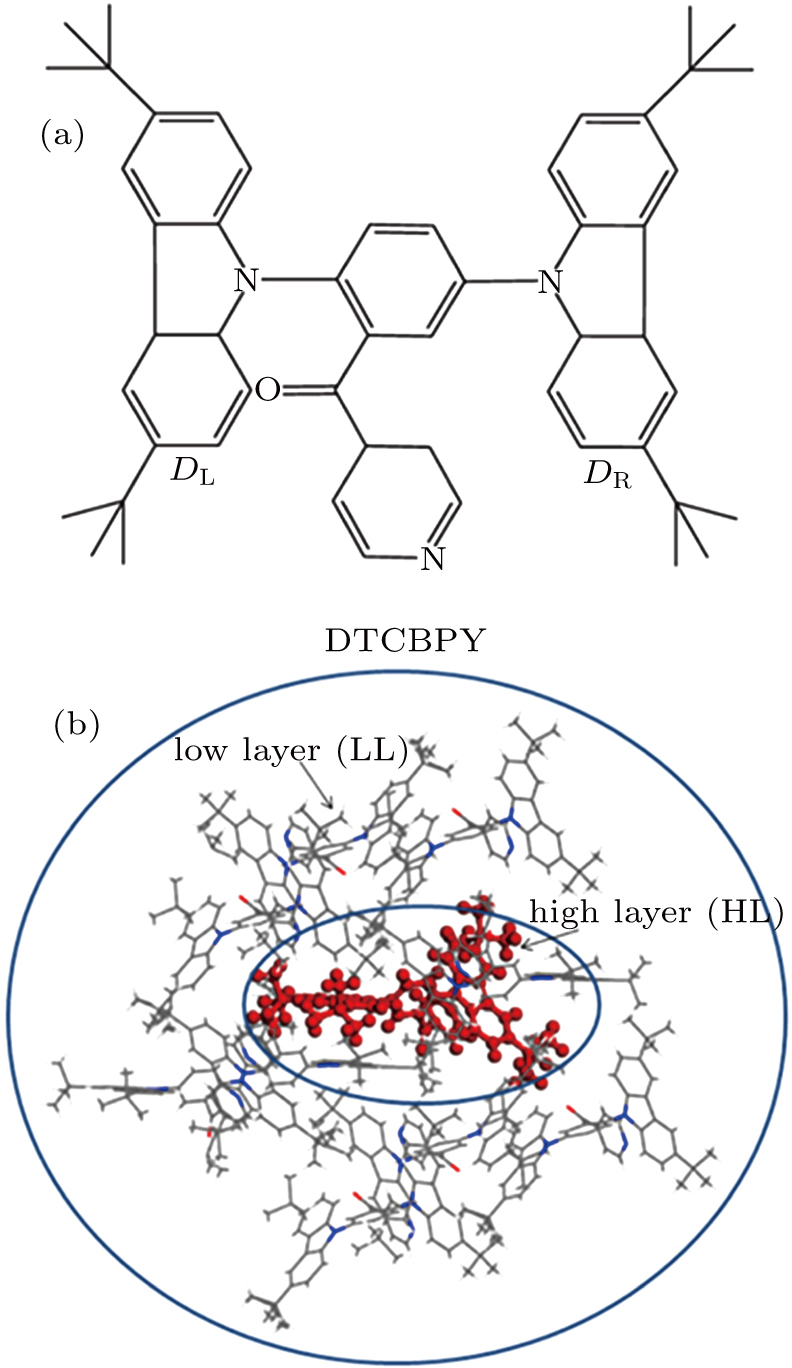 | Fig. 1. (color online) (a) Geometry structure of the studied molecule DTCBPY. (b) ONIOM model: the centered C configuration is treated as a high layer and the surrounding configurations are fixed as a low layer. |
2. Computational methods
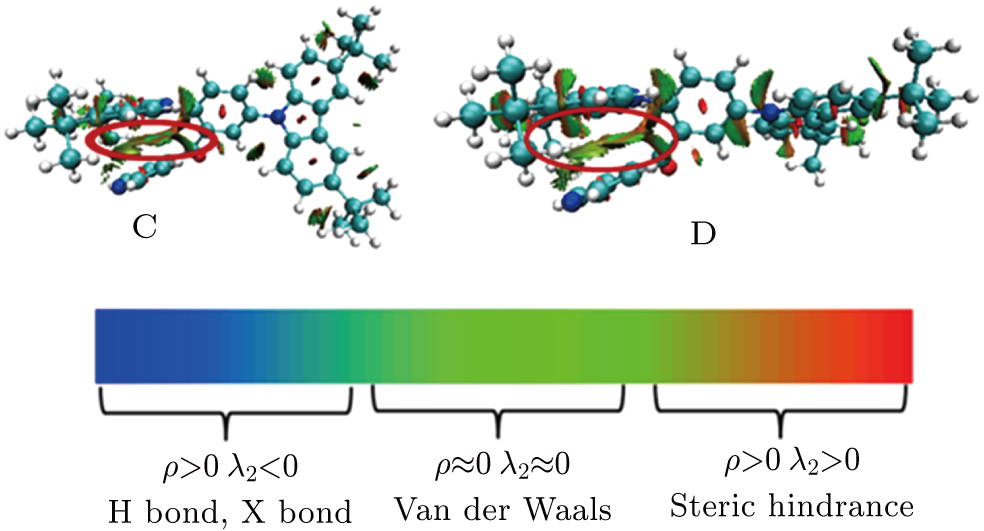
where ρ(r) is the electronic density. Through analysis of the RDG values, one can easily distinguish the area of near nuclei, near chemical bonds region, weak interacting regions and the edges of the molecule. This method can also highlight the areas of π–π intramolecular interaction in the system and help to visualize the region that is associated with the intramolecular π–π interaction.[29]
In this paper, the density functional theory (DFT) is used to perform the optimization and electronic structure calculation of the molecule in ground state. The time-dependent density functional theory (TD-DFT) is employed to investigate the properties of the excited states. In the calculations, the pbe0-1/3 exchange functional and the 6-31G(d) basis set are used. The polarizable continuum model (PCM) is used to take into account the solvent effect in all simulations.[24,25]
The photoluminescence quantum yield (PLQY) is determined by the radiative rate (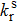
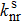
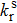
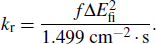 |
Here f is the oscillator strength without dimension and ΔEfi in units of wavenumber (cm−1) is the energy difference between the initial state and the final state.[26] 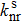
In order to visualize the intramolecular interaction between the donor and the acceptor, the reduced density gradient (RDG) method is adopted. The RDG formula is written as
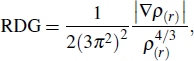 |
In experiment, the PLQY of the molecule in toluene is 30.3%, while it reaches up to 91.4% in solid state. It means that the luminescence efficiency of the molecule is greatly enhanced in the aggregation state. To explain this phenomenon, the combined quantum mechanics and molecular mechanics (QM/MM) method is adopted to study the electroluminescent properties of the DTCBPY molecule by considering the environment effect in aggregation state.[28] The initial structure is obtained from the x-ray crystal structure detected experimentally.[19] The QM/MM calculation is realized with the ONIOM method in Gaussian 09 program.[24] In this model, two “layers” are constructed, as shown in Fig.
3. Results and discussion
3.1. Molecular geometric structure
where p is the proportion, i represents the configuration, ni is the number of molecules in the i-th configuration, and E is the energy of the configuration. T is the temperature (in units of K), and R is the ideal gas constant. Q is the partition function. By using the relative energies of different conformations, the Boltzmann distribution formula can be rewritten as
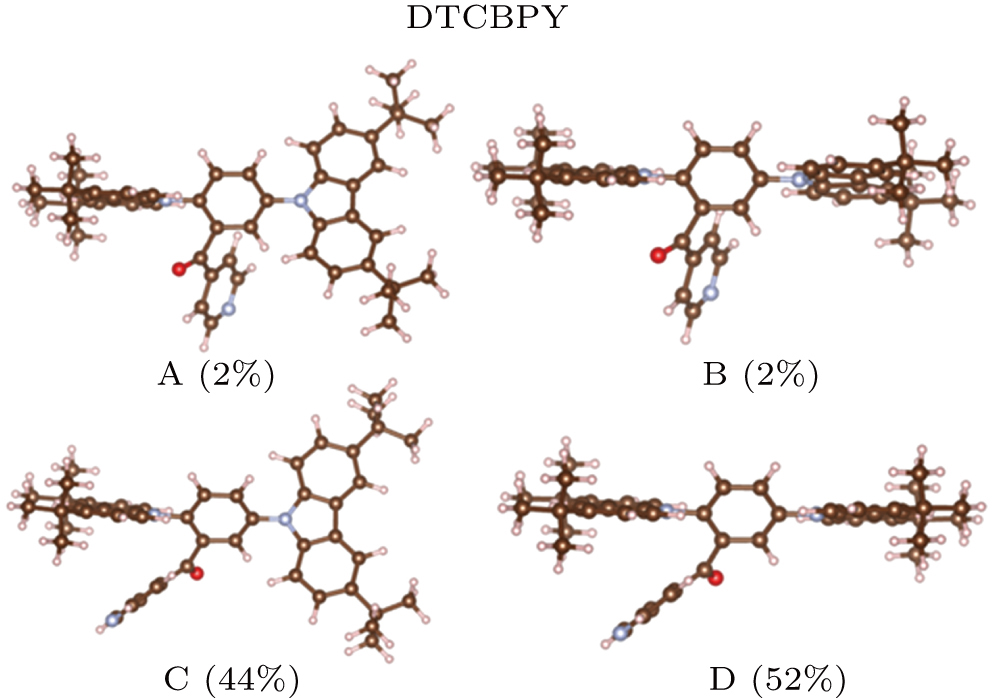
The DTCBPY molecule consists of two donors and one acceptor, which possesses a flexible configuration, as shown in Fig.
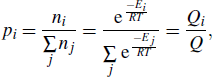 |
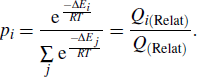 |
Here ΔEi is the relative value of conformation i, and the values are shown in Table
 | Fig. 2. (color online) Four configurations of the DTCBPY and the proportion ratios of the configurations. |
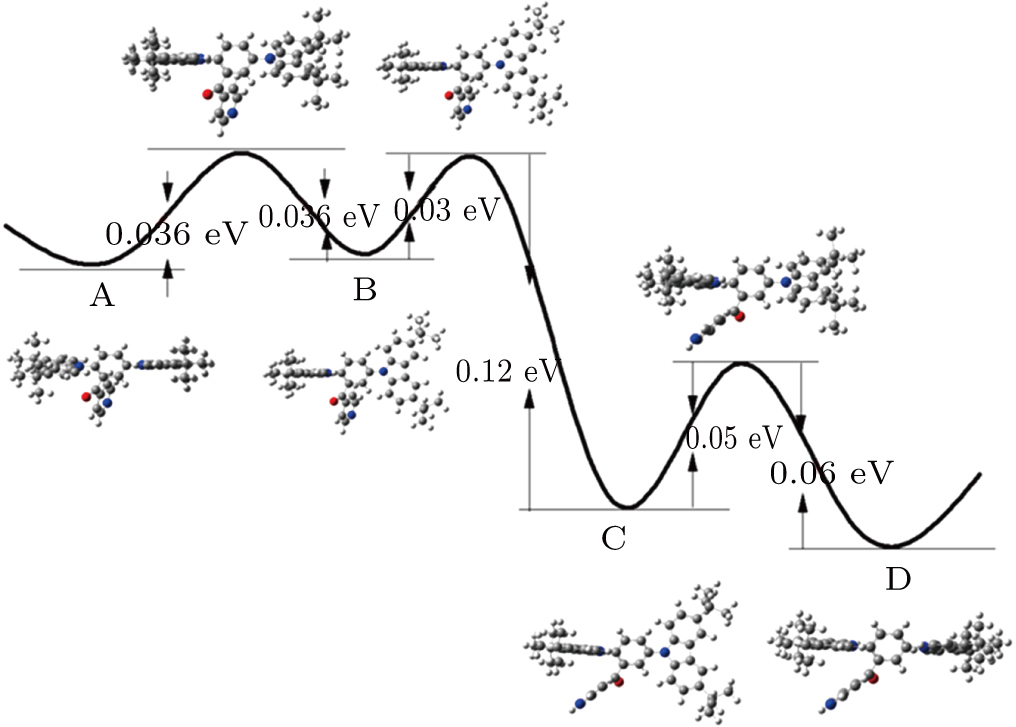
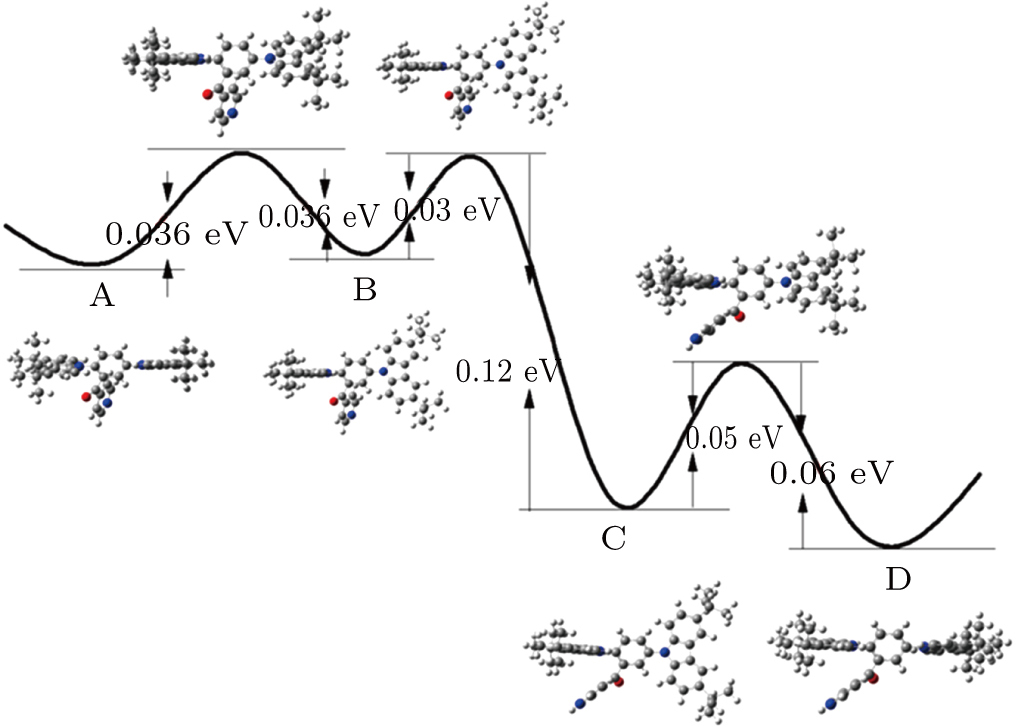 | Fig. 3. (color online) Potential energy surface and energy barrier of the molecule. The geometric structure of the molecule in each point is also presented. |
| Table 1.
Absorption wavelength, emission wavelength, oscillator strength, radiative rate, stokes shift (λshift), and energy gap for the four configurations. . |
| Table 2.
Relative energy of the four conformations and their proportions. . |
Based the analysis above, the configurations of C and D have the most possible configurations. From the configurations of C and D, we can see that the acceptor group is close to one donor group, thus we deduce that there is intramolecular π–π interaction between them. To visualize the intramolecular interaction more intuitively, the RDG method is used (shown in Fig.
 | Fig. 4. (color online) Intramolecular π–π interaction between the acceptor and the donor nearby described by the RDG method. The weak interacting area is marked with the red circle. |
3.2. Excited state property and energy gap
where ϕH and ϕL represent the wave functions of the HOMO and LUMO, respectively. For TADF molecules, an endothermic up-conversion process from T1 to S1 state can be thermally activated when ΔEst is small. The small ΔEst can be achieved by decreasing the overlap between HOMOs and LUMOs. The f is determined by the transition dipole moment μ which can be written as
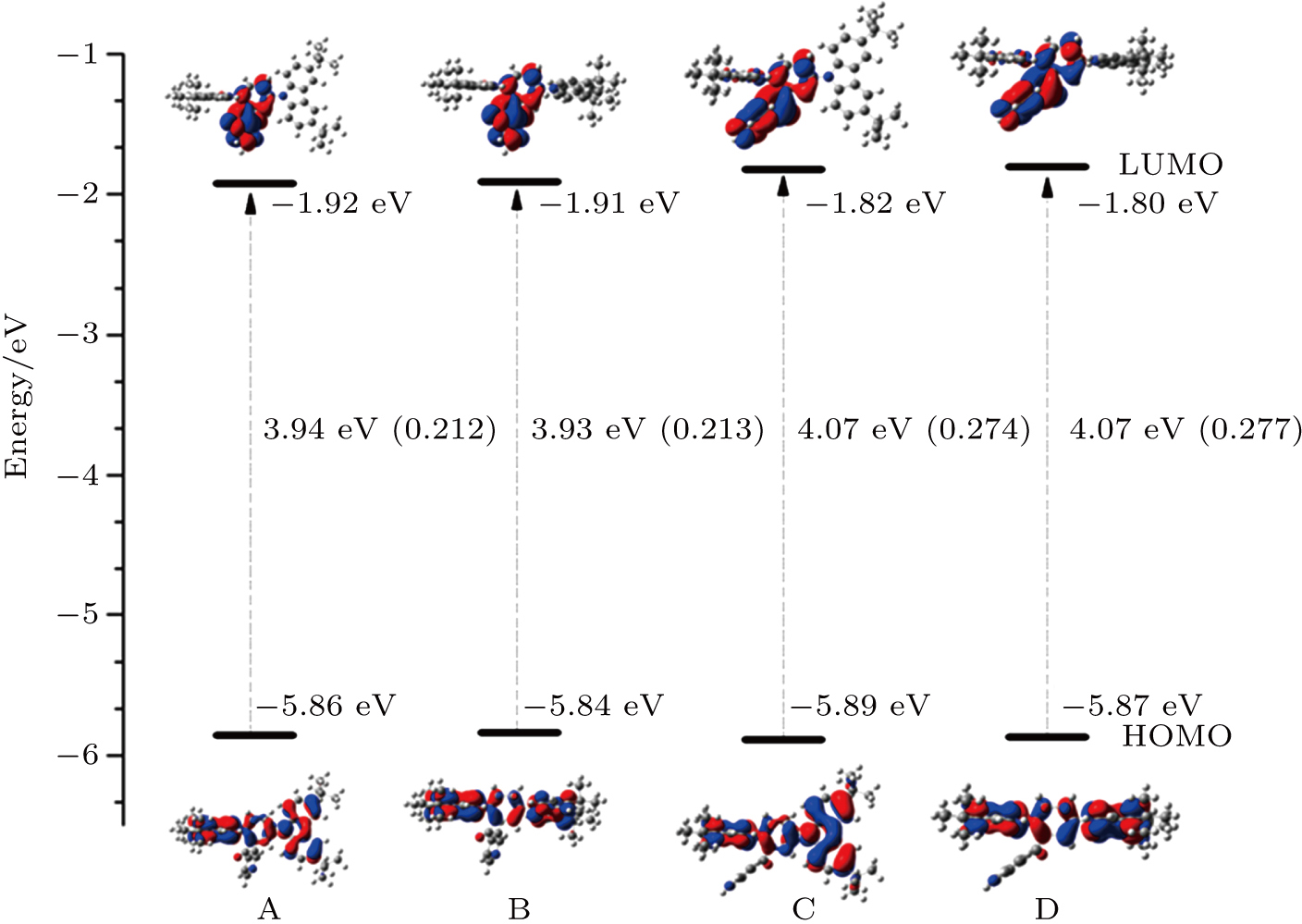
The electron distributions of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) as well as their energy levels for four configurations are shown in Fig. 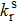
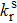
 |
 | Fig. 5. (color online) Distributions of frontier molecular orbitals of the configurations. The energy levels of HOMOs and LUMOs as well as their energy gaps are also shown. The overlaps between HOMOs and LUMOs are listed in the bracket. |
Here K is the exchange energy, and it can be calculated by the formula
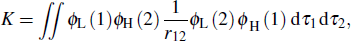 |
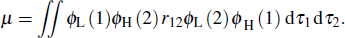 |
One can see that the overlap between HOMO and LUMO can also influence μ. Thus f will also be determined by the HOMO–LUMO overlap. The larger the overlap, the bigger the f. As a result, the TADF molecules should have a proper overlap between HOMOs and LUMOs, which can be regulated by adjusting the intramolecular interaction. Based on the analysis above, we find that the intramolecular π–π interaction between the acceptor and the donor can effectively enhance the light-emitting efficiency, which may help us to design highefficient light-emitting materials.
3.3. Influence on the non-radiative rate
Reorganization energy (λ) can be expressed as a summation of the contributions from normal mode (NM) relaxation in the harmonic oscillator approximation[27]
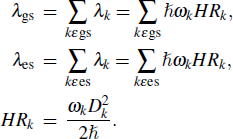 |
In the formula, HRk represents the Huang–Rhys factor for the k-th mode, and Dk is the displacement for mode k between the equilibrium geometries of S0 and S1. The maximum Huang–Rhys factor (HRmax) and the corresponding λ are listed in Table 
| Table 3.
Reorganization energy λ and the maximum Huang–Rhys factor HRmax for four configurations. . |
Further, the QM/MM method is used to study the change of the dihedral angles of the molecule in ground state and excited state in solution and in solid state (as listed in Table 
| Table 4.
Dihedral angles between the left (right) donor and the acceptor (DL and DR respectively) for the molecule in ground state and S1 state. The differences are also shown. . |
4. Conclusion
We have studied a TADF molecule named DTCBPY with the DFT and TD-DFT methods. Through relax scanning, we find that the molecule has four different conformations, and the configuration C is consistent with the molecule structure detected in the experiment. Our theoretical results also indicate that both C and D have the most possible configurations for DTCBPY. By using the RDG function, we confirm that there is significant π–π interaction between the acceptor and the donor groups nearby in C and D. By analysis of the Huang–Rhys factor and the reorganization energy, we deduce that the intramolecular interaction can induce a smaller non-radiative rate and a high PLQY. In addition, the QM/MM method is used to study the structural changes of the molecule between the ground state and the S1 state in the solid state. Compared with the dihedral angle change in a solvent, the dihedral angle change in a solid state decreases obviously, which may result in suppressed non-radiative transition and enhanced luminous efficiency. Based on our calculation results, we deduce that the molecule may have the properties of aggregation induced enhanced emission. Our theoretical research provides valuable information for the design of highly-efficient organic light-emitting molecules.
Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] | |
| [32] |