

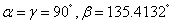
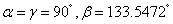
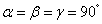

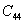
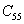
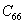
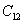


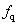


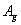
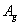


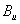
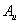



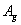
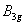
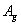
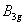
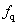
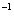

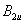
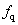
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Crystal structures and electronic properties of solid fluorine under high pressure
Cite this Article
Lv Qianqian, Jin Xilian, Cui Tian, Zhuang Quan, Li Ying, Wang Youchun, Bao Kuo, Meng Xing. Crystal structures and electronic properties of solid fluorine under high pressure. Chinese Physics B, 2017, 26(7): 076103 
Permissions
Crystal structures and electronic properties of solid fluorine under high pressure
† Corresponding author. E-mail:
Abstract
As the previously proposed structures of C2/m and C2/c possess similar enthalpies and x-ray diffraction patterns, the space group of fluorine at ambient pressure is in controversy. We successfully obtain its thermodynamically stable low-pressure phase, which shares the same structure as the earlier known C2/c. Further investigations on phonon spectra reveal the instability of the C2/m structure with imaginary frequency in the Brillouin zone and confirm the dynamically stable property of the C2/c structure at the same time. Compressing fluorine up to 8 GPa, the C2/c phase is found to undergo a phase transition to a new structure with a space group of Cmca. Electronic energy band structures indicate the insulating feature of C2/c and Cmca with no bands across the Fermi level. The infrared (IR) and Raman spectra of C2/c and Cmca at selected pressures are calculated to provide useful information to future experiments.
1. Introduction
As a long-lived issue in physics and chemistry, high-pressure properties of diatomic molecules, such as H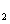
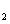
Fluorine, the first of the halogens and the last of the first-row diatomic elements, has many characteristics similar to hydrogen. On the other hand, elemental fluorine is also an abundant element in the universe and it is a system of particular interest and exhibits many unusual physical and chemical properties by virtue of the strongest oxidability and electronegativity. The fluorine-related systems have also been studied extensively, for example, BaF
The crystal structure of 
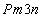
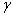

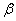
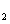
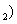

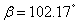
Besides, the high-pressure properties of solid fluorine have not been sufficiently covered, thus the structures of crystal fluorine in a wide pressure range of 0–100 GPa are explored by using first-principles calculations based on the density functional theory (DFT)[46,47] in this paper. In this work, the thermodynamically stable phase C2/c at low-pressure is obtained by us, which possesses the same structure as earlier known C2/c.[40] Our high precision calculations on enthalpies of C2/m and C2/c fluorine through the VASP code suggest that the C2/c phase is slightly more energetically stable than C2/m at low pressure. Further investigations indicate that C2/m has imaginary phonon frequencies, while C2/c establishes dynamical stability. Theoretically, C2/c is determined to be the better candidate of solid fluorine at ambient pressure. At about 8 GPa, C2/c transforms to a new high-pressure phase Cmca proposed by us. Cmca remains intact at the pressure of 100 GPa, which is not similar to the other halogen molecules that readily exhibit molecular dissociation. The electronic properties demonstrate that both C2/c and Cmca are insulators. The nonmetallic property of Cmca fluorine is different from that of the other halogen crystals with the prototypical structure of Cmca.
2. Calculation methods
We use the ELocR[48] code to predict the structure of fluorine under high pressure. The energetic and electronic structure calculations are performed with the Vienna ab initio simulation package (VASP)[49] within density-functional theory using the Perdew–Burke–Ernzerh (PBE) of generalized gradient approximation (GGA).[50] The 2s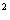
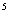
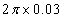
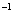
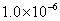
3. Results and discussion
The crystal structures of fluorine are explored through ELocR, the calculated enthalpy difference 

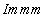
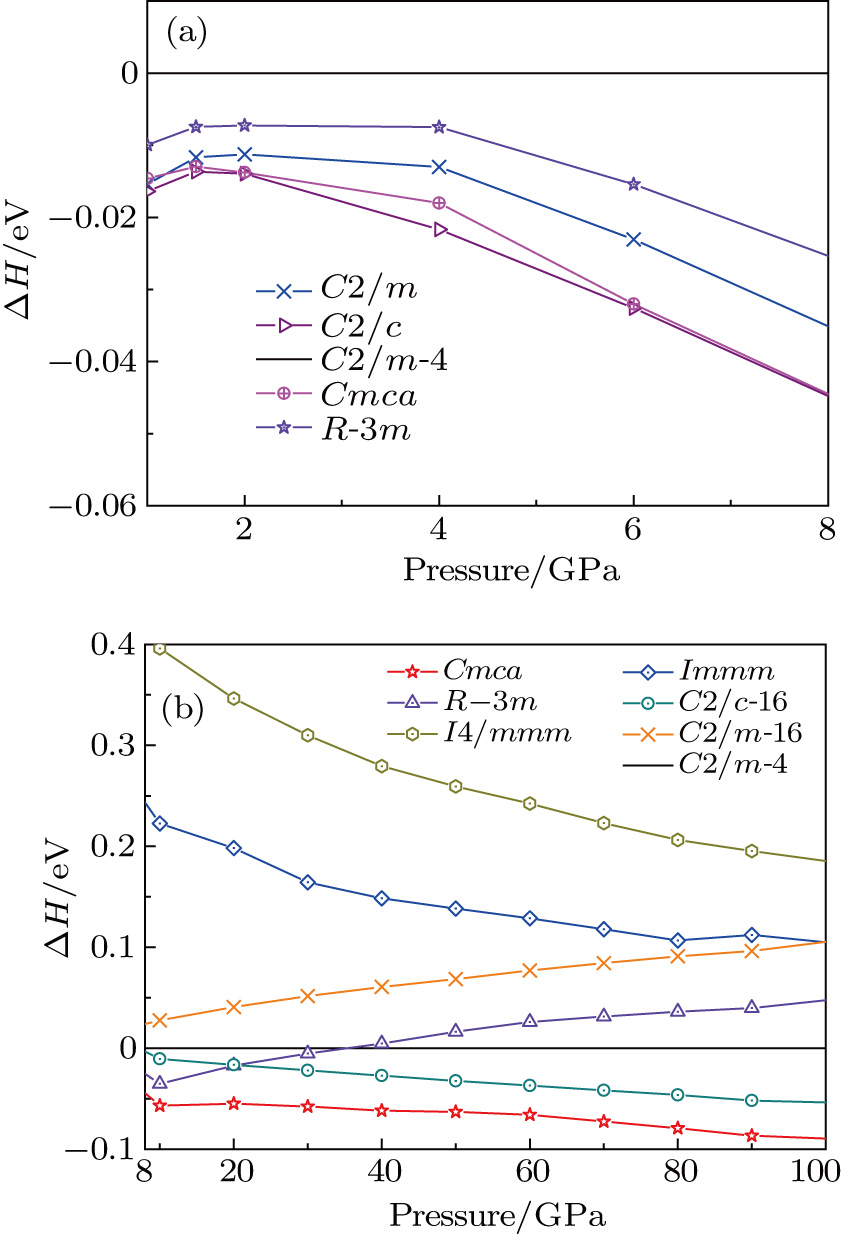 | Fig. 1. (color online) Calculated enthalpy per fluorine atom relative to C2/m-4 as a function of pressure in the pressure ranges of (a) 1–8 GPa and (b) 8–100 GPa. |
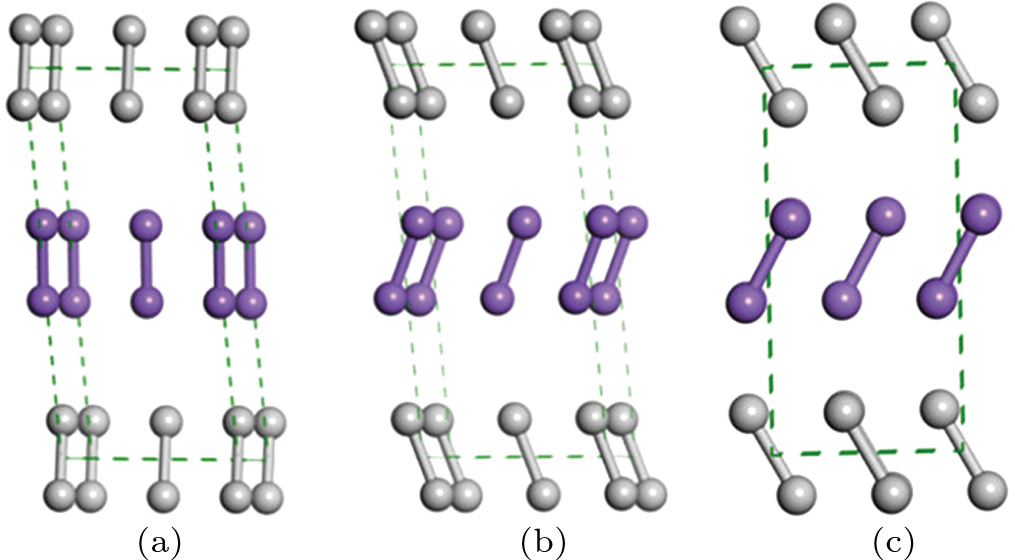
The favored structures of solid fluorine obtained in this work are illustrated in Fig.
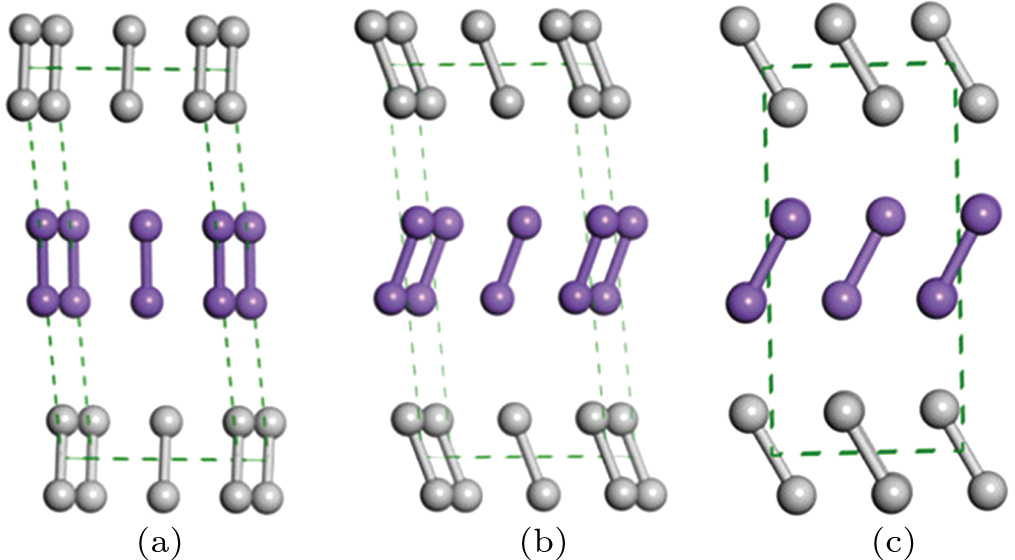 | Fig. 2. (color online) The structures of fluorine at selected pressures: (a) C2/m at 0 GPa, (b) C2/c at 0 GPa, (c) Cmca at 20 GPa. Different color fluorine molecules represent different kinds of molecules. |
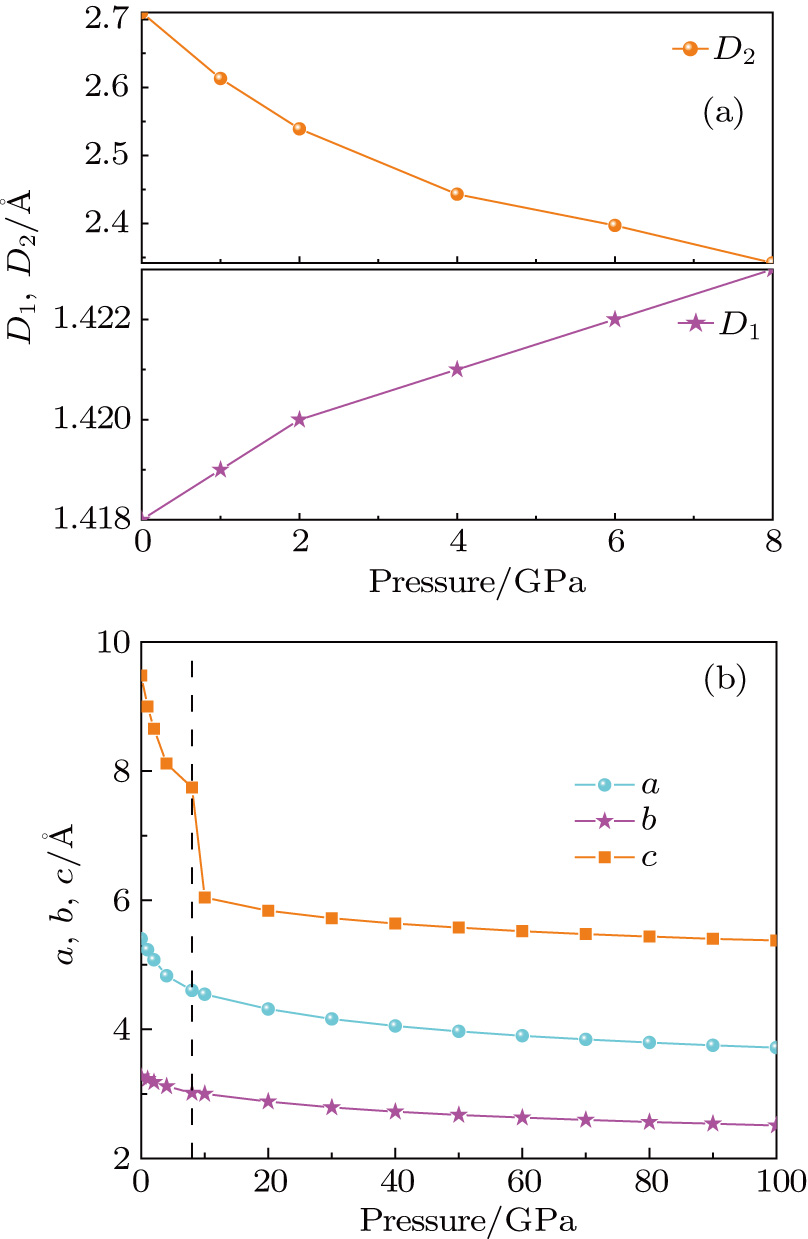
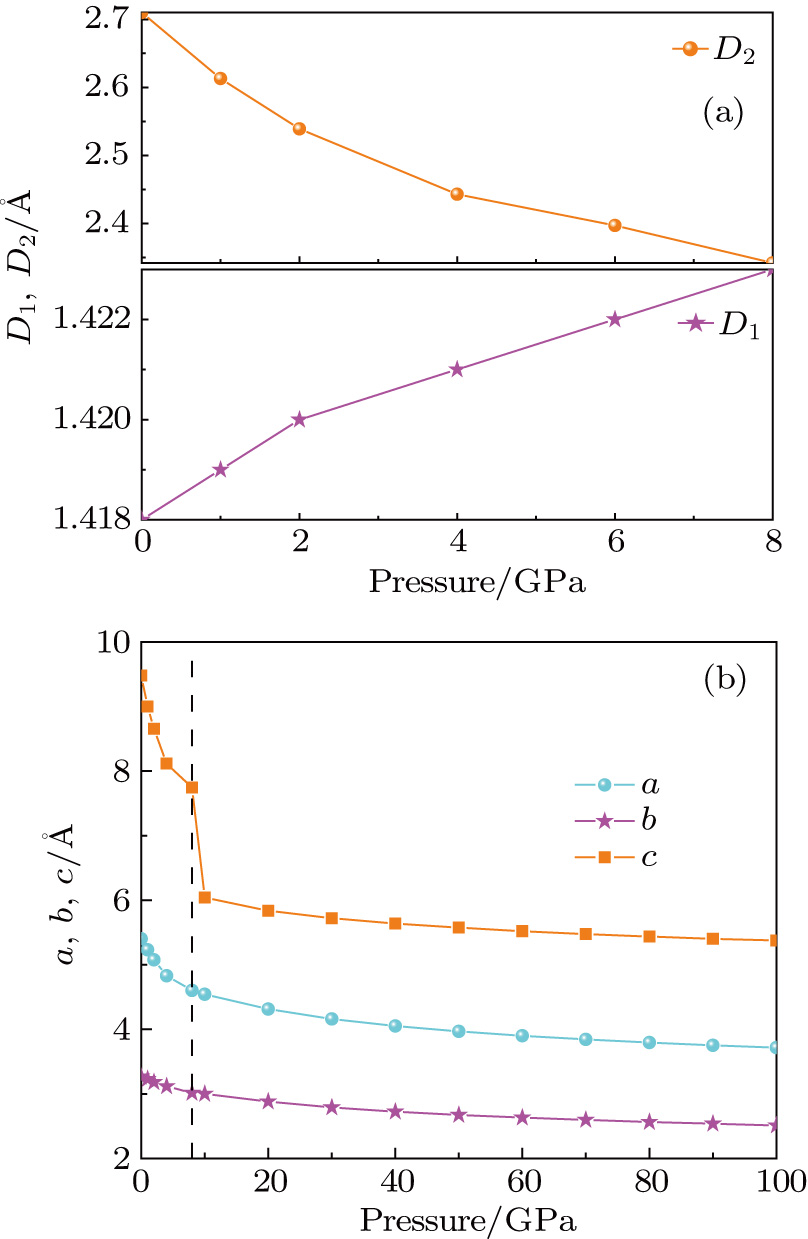 | Fig. 3. (color online) Neighboring distances in the C2/c phase and lattice parameters of crystal fluorine plotted as a function of pressure: (a) neighboring distances, 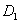 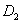 |
| Table 1.
Optimized structural parameters for solid fluorine at selected pressures. . |
The phenomena are also observed in the C2/c structure of solid hydrogen.[52] The differences between hydrogen and fluorine are that the C2/c fluorine has an equivalent intramolecular bond length, while the C2/c hydrogen possesses two different bond lengths. Moreover, the shorter intramolecular distance and the typical intermolecular distance of solid hydrogen reach equality at 600 GPa upon compression and the hydrogen molecules start to dissociate. However, the intermolecular distance of the C2/c fluorine remains longer than the intramolecular bond length all the time. With increasing pressure, the C2/c fluorine transforms into Cmca without any dissociation. The lattice parameters as a function of pressure in solid fluorine are presented in Fig.
The lattice of Cmca is orthorhombic with axes a = 4.3115, b = 2.8812, c = 8.8082 at 20 GPa. The cyrstallographic sites of the eight atoms in the conventional cell are in the position 8f (x = -0.5000, y = 0.3799, z = 1.39328). The crystal fluorine of the Cmca phase also has a layered structure and the distribution of fluorine molecules is similar to that of C2/c structure as shown in Fig.
For a stable crystal structure, mechanical stability is a necessary condition and it requires the strain energy to be positive, i.e., the whole set of elastic constants 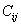
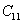
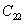
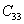
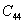
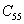
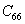

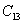
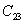
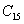



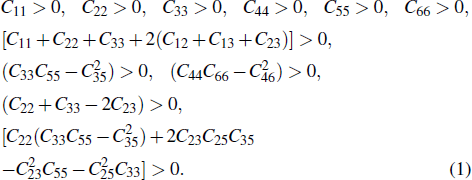 |
For an orthorhombic crystal, the independent elastic stiffness tensor consists of nine components 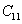
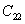
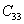
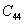
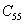
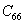

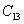
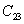
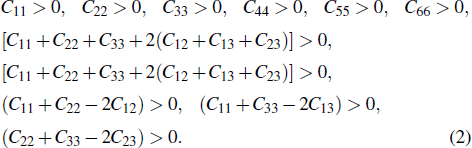 |
The elastic stiffness constants 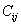
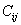
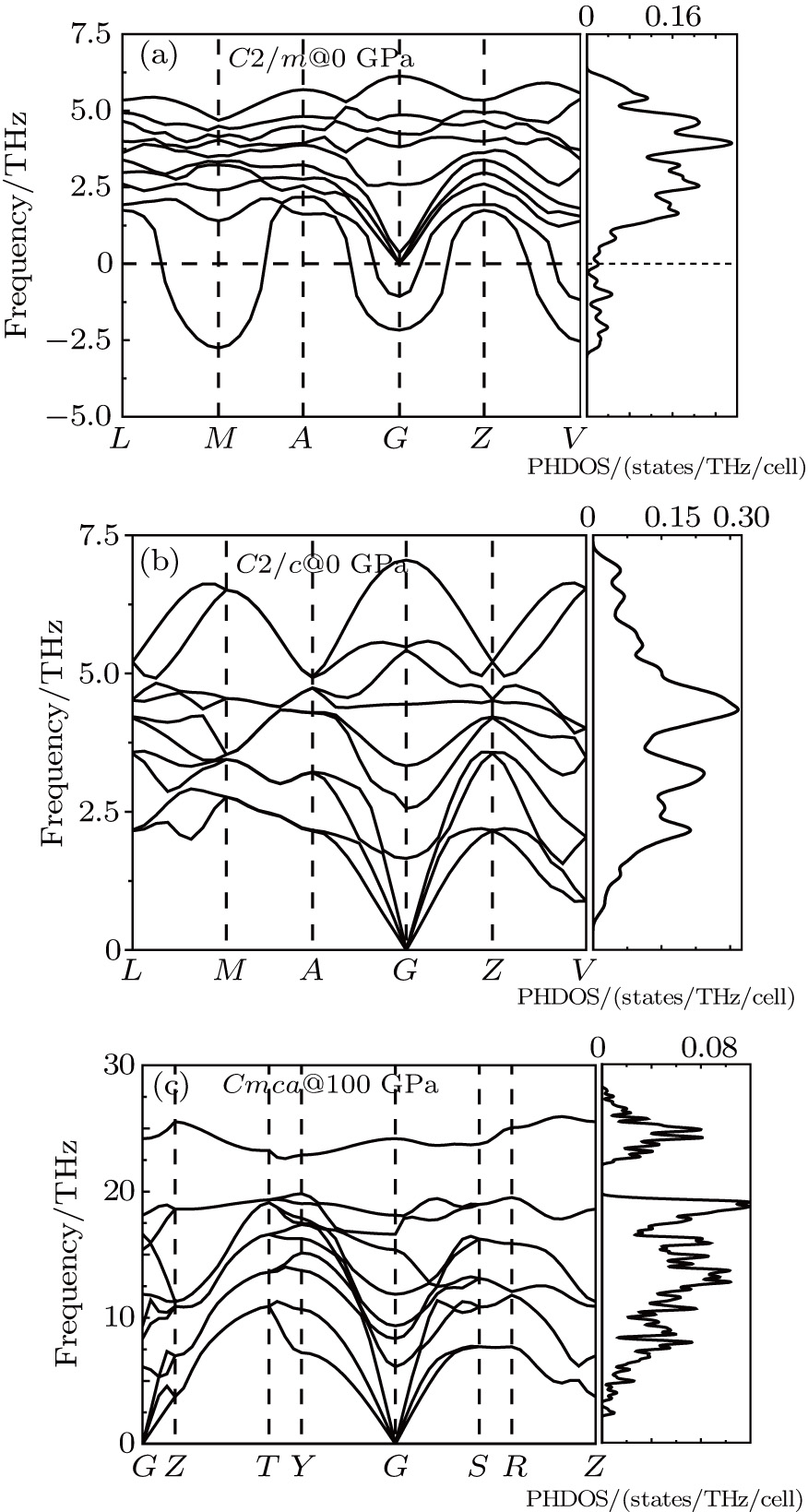 | Fig. 4. Calculated phonon dispersion curves for (a) C2/m at 0 GPa, (b) C2/c at 0 GPa, and (c) Cmca at 100 GPa. |
| Table 2.
The calculated elastic stiffness coefficients (in GPa) of C2/c at ambient pressure and Cmca at 100 GPa. . |
It is worth mentioning that the x-ray powder diffraction experiment carried out on solid fluorine provided two candidates with the space groups of C2/m and C2/c. As geometry optimization is applied to the controversial structures of C2/m and C2/c, the calculated lattice parameters of C2/m phase (a = 5.4702, b = 3.2316, c = 10.0675) and C2/c phase (a = 5.4009, b = 3.2657, c = 9.4781) at ambient pressure are comparable with the experimental values (a = 5.50, b = 3.28, c = 10.01). Both C2/m and C2/c structures are monoclinic, the fluorine molecule distributions are analogous to each other, except they differ slightly in their molecular orientations as shown in Figs.
Although small different enthalpies between C2/m and C2/c crystals exist as mentioned above, C2/c is more energetically stable than C2/m at low pressure. Further investigations on the lattice dynamic stability reveal the dynamical stability of C2/c comparing the instability of C2/m with imaginary frequency in the vicinity of high symmetry points of M, G, and V in the Brillouin zone, as shown in Figs.
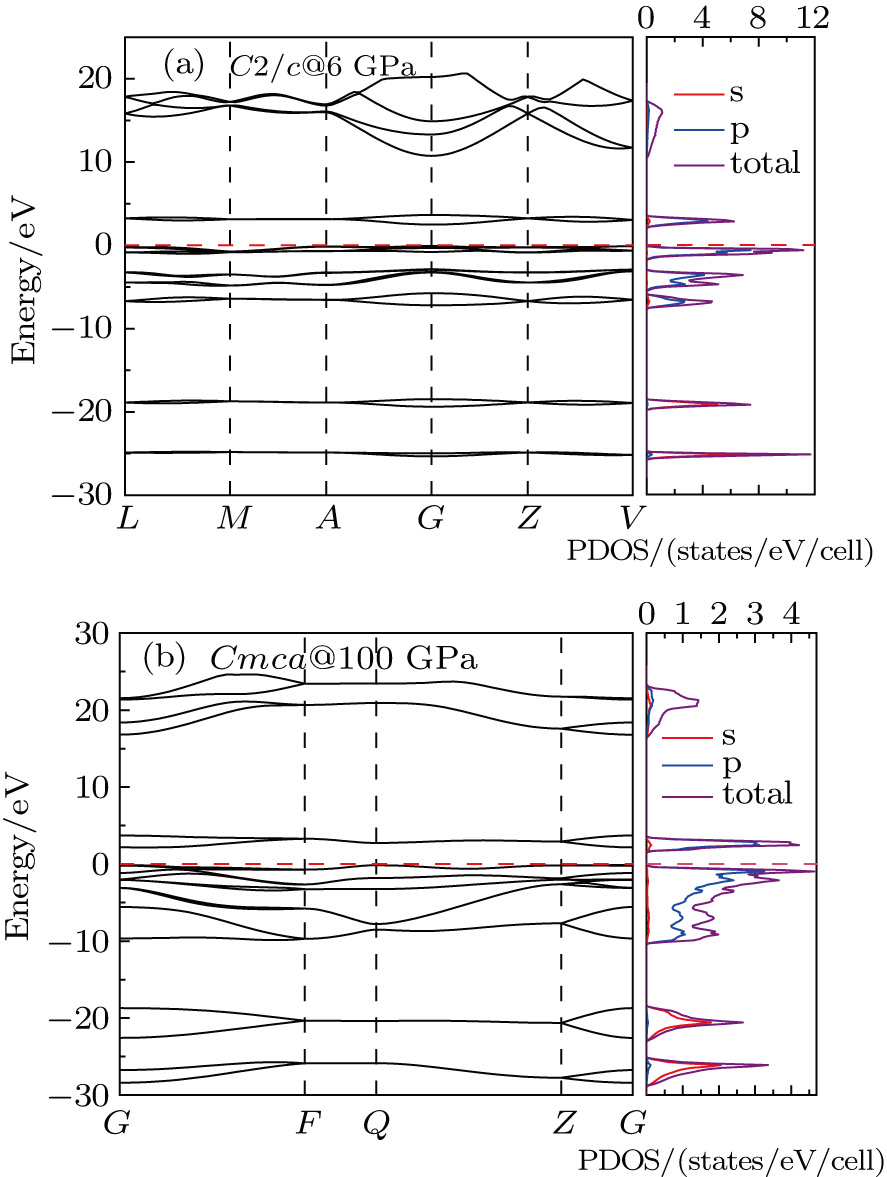
To uncover the electronic properties of the C2/c and Cmca phases, the electronic band structure and the corresponding density of states (DOS) of C2/c at 6 GPa and Cmca at 100 GPa are calculated and illustrated in Fig.
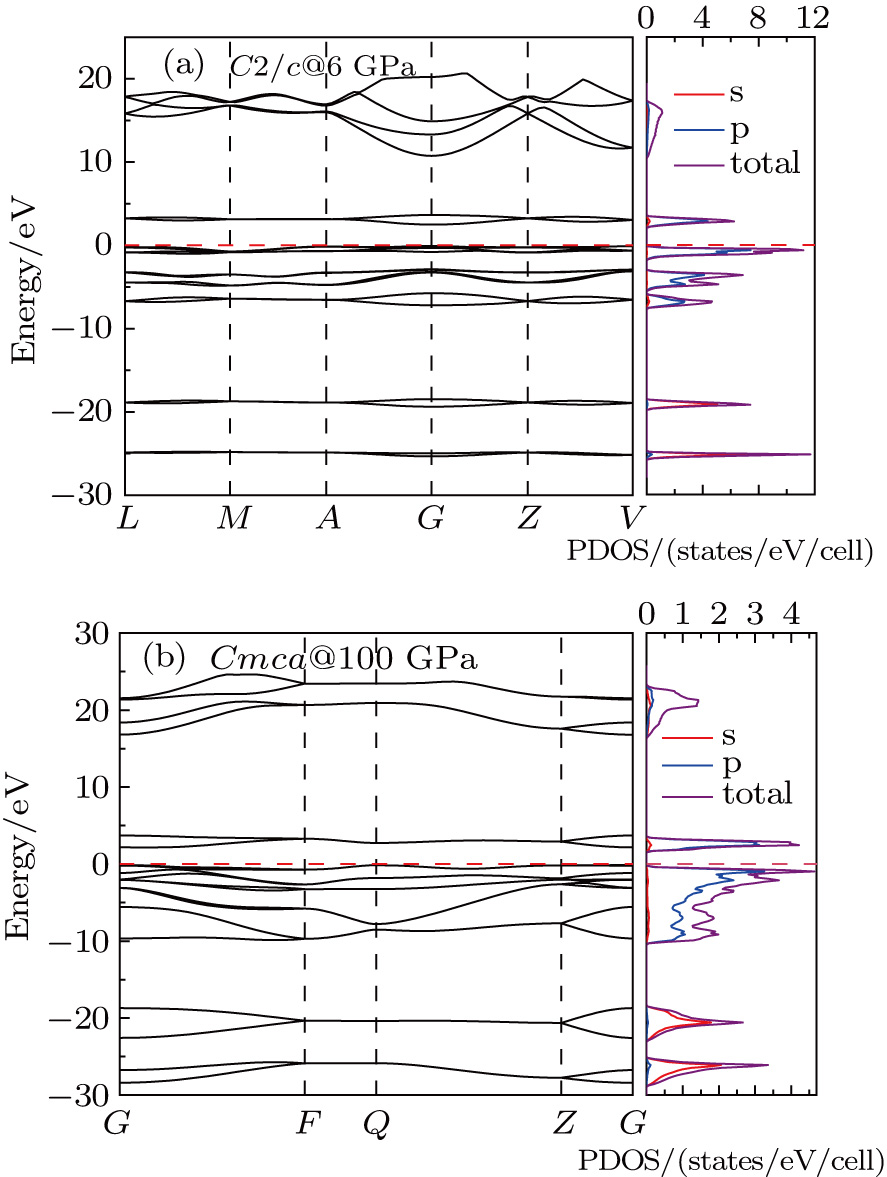 | Fig. 5. (color online) The calculated electronic band structure and the corresponding density of states (DOS): (a) C2/c at 6 GPa, (b) Cmca at 100 GPa. |
Allowing for less high-pressure experimental data of crystal fluorine, we calculate the IR and Raman spectra of C2/c and Cmca at 1 GPa and 20 GPa, respectively, as described in Table 
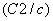

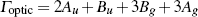

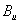
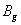


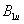
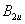
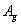
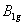
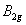
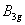
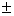
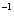


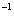
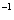


| Table 3.
Computed IR/Raman active frequencies for the C2/c and Cmca structures at 1 GPa and 20 GPa, respectively. Here R and I stand for Raman and IR active, vm and |
4. Conclusion
In summary, high precision calculations on enthalpies of the controversial structures of C2/m and C2/c fluorine through the VASP code suggest that theC2/c phase is slightly more energetically stable than C2/m at low pressure. Further investigations on phonon spectra reveal the instability of the C2/m structure and confirm the dynamically stable property of the C2/c structure. Theoretically, C2/c is determined to be the better candidate of solid fluorine at ambient pressure. Under compression, the C2/c phase transforms to a new structure Cmca at about 8 GPa. The Cmca phase is superior to the other structures in energy in the pressure range of 8–100 GPa. Moreover, Cmca remains intact at the pressure of 100 GPa, which is not similar to the other halogen molecules. The electronic energy band structures indicate that both C2/c and Cmca structures are insulators at the selected pressures.
Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] | |
| [32] | |
| [33] | |
| [34] | |
| [35] | |
| [36] | |
| [37] | |
| [38] | |
| [39] | |
| [40] | |
| [41] | |
| [42] | |
| [43] | |
| [44] | |
| [45] | |
| [46] | |
| [47] | |
| [48] | |
| [49] | |
| [50] | |
| [51] | |
| [52] | |
| [53] | |
| [54] | |
| [55] | |
| [56] |