

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Geometric stability and electronic structure of infinite and finite phosphorus atomic chains
Cite this Article
Qiao Jingsi, Zhou Linwei, Ji Wei. Geometric stability and electronic structure of infinite and finite phosphorus atomic chains. Chinese Physics B, 2017, 26(3): 036803 
Permissions
Geometric stability and electronic structure of infinite and finite phosphorus atomic chains
† Corresponding author. E-mail:
Project supported by the National Natural Science Foundation of China (Gant Nos. 11274380, 91433103, 11622437, and 61674171), the Fundamental Research Funds for the Central Universities, China, and the Research Funds of Renmin University of China (Grant No. 16XNLQ01). Qiao was supported by the Outstanding Innovative Talents Cultivation Funded Programs 2016 of Renmin University of China.
Abstract
One-dimensional mono- or few-atomic chains were successfully fabricated in a variety of two-dimensional materials, like graphene, BN, and transition metal dichalcogenides, which exhibit striking transport and mechanical properties. However, atomic chains of black phosphorus (BP), an emerging electronic and optoelectronic material, is yet to be investigated. Here, we comprehensively considered the geometry stability of six categories of infinite BP atomic chains, transitions among them, and their electronic structures. These categories include mono- and dual-atomic linear, armchair, and zigzag chains. Each zigzag chain was found to be the most stable in each category with the same chain width. The mono-atomic zigzag chain was predicted as a Dirac semi-metal. In addition, we proposed prototype structures of suspended and supported finite atomic chains. It was found that the zigzag chain is, again, the most stable form and could be transferred from mono-atomic armchair chains. An orientation dependence was revealed for supported armchair chains that they prefer an angle of roughly 35°–37° perpendicular to the BP edge, corresponding to the [110] direction of the substrate BP sheet. These results may promote successive research on mono- or few-atomic chains of BP and other two-dimensional materials for unveiling their unexplored physical properties.
1. Introduction
With respect to the minimization of electronic and optoelectronic devices, the dimensionality of devices has been reducing from three-dimension to two-dimension (2D) and eventually to one-dimension (1D).[1–4] The ultimate small-size 1D devices are expected to be comprised of one or few-atoms width atomic chains which were realized in graphene, BN, and transition metal dichalcogenides (TMDs) by knocking out atoms from 2D nanosheets under electron beam irradiation.[5–9] These atomic chains were found of striking properties including excellent electric transport, high thermopower, and ultra-strong mechanical strength. Black phosphorus (BP), the most stable allotrope of phosphorus at ambient conditions, has attained tremendous attention since its rediscovery as a high-mobility, high thermo-power, anisotropic, and tunable direct bandgap semiconductor with dichroism optical response in 2014.[10–13] It is the very first elemental semiconductor in 2D layered materials with its bandgap covering near-infrared to visible light region.[11,14] It would be thus interesting if BP atomic chains could be realized in experiment and if these chains have any striking physical properties. Both of these questions are unknown and subject to further investigations.
In light of the experimental protocols adopted in the previous experiments,[15–17] a few-atom width ribbon is a precursor of mono-atomic chains, which could be fabricated by creating two parallel line defects apart by a few atoms. Recently, a theory predicted that a chain-like vacancy may form in BP nanosheets under electron irradiation, while similarline defects were observed in experiment during the period of summarizing this work.[18] Given these chain-like vacancies, the exact atomic structure of BP chains, the thermal stability and electronic structures of them are still unclear.
Here, we employ density functional theory (DFT) calculations to consider the likely structures of mono- and dual-atomic infinite and encapsulated finite BP chains. Three categories of mono-atomic infinite chains, namely, linear (L), armchair (AC), and zigzag (ZZ) chains, in addition with three associated categories of dual-atomic infinite chains, were considered in our calculations. It was found that zigzag chains are always energetically stable in both mono- and dual-atomic chains while linear chains are of the highest energies. Phase transitions among them were revealed relevant with the linear density of P atoms along the chain, as elucidated in a phase diagram. Electronic structures of these six chains were also discussed that a likely band inversion with spin–orbit coupling (SOC) was found in the infinite ZZ chain. Geometry and stability of finite chains were considered in models of a chain encapsulated in two BP half-sheets with and without the support from BP sheets underneath, which represent the most likely experimental cases.
2. Methods
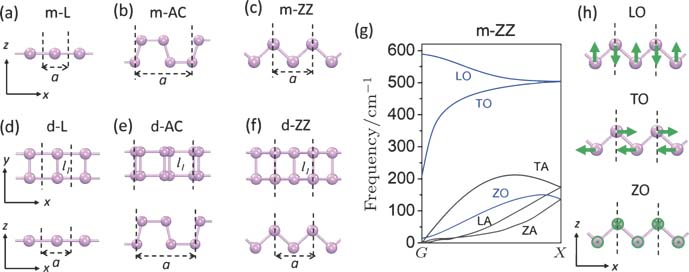
Density functional theory calculations were performed using the generalized gradient approximation for the exchange–correlation potential, the projector augmented wave method,[19,20] and a plane wave basis set as implemented in the Vienna ab-initio simulation package (VASP)[21] and the quantum espresso (QE)[22] code. Density functional perturbation theory (DFPT)[23] was employed to calculate phonon dispersion relations using QE. In geometry optimization, van der Waals interactions were considered at the van der Waals density functional (vdW-DF)[24,25] level with the optB86b functional for the exchange potential (optB86b-vdW),[26,27] which was found more suitable than other vdW-DF functionals for modeling mechanical properties and interlayer coupling strength of 2D materials.[28–33] In terms of infinite chains, the energy cutoff for the plane-wave basis was set to 700 eV. A supercell containing only one (two) P atoms with vacuum regions of ∼ 20 Å in the y and z directions was used to model the infinite linear (dual) atomic chain (Fig.
 | Fig. 1. (color online) Geometric stucutres of six representative 1D infinite atomic chains: (a) mono-linear chain (L), (b) mono-armchair chain (AC), (c) mono-zigzag chain (ZZ), (d) dual-linear chain (d-L), (e) dual-armchair chain (d-DB), (f) dual-zigzag chain (d-ZZ). Lattice constant a is marked on each configuration. Bond lengths l1 of dual-chains are labeled in panels (d)–(f). (g) Phonon dispersion relation of the m-ZZ chain. (h) Vibrational displacements for three optical modes of the m-ZZ chain. |
3. Results and discussion
3.1. Stability of infinite P chains
Figures
3.2. Structural phase transition among infinite chains
These six infinite chains are transferable by stretching or compressing them along the infinite direction. It thus requires a parameter describing the relationship between length and energy. Here, we introduce a geometric parameter (n) which represents the length, in units of Å, of each P atom in a chain. The n values, the inversion of linear density of P atoms, are 2.13 Å, 1.57 Å, and 1.36 Å for m-L, m-ZZ, and m-AC, respectively, while those n values for the dual-atomic chains are almost one-half of the corresponding values of the mono-atomic chains, i.e., 1.15 Å, 0.83 Å, and 0.70 Å, respectively. A phase diagram was plotted by recording energy evolution of different chains during elongation and contraction, as shown in Fig.
 | Fig. 2. (color online) Phase diagram of infinite atomic chains shows the relationship between energy and the length per P atom (n). Cube, circle, and triangle are correspondind to linear, zigzag, and armchair chains, respectively. Mono- and dual-atomic chains are indicated by solid and hollow shapes. |
3.3. Electronic structures of infinite P chains
Figures
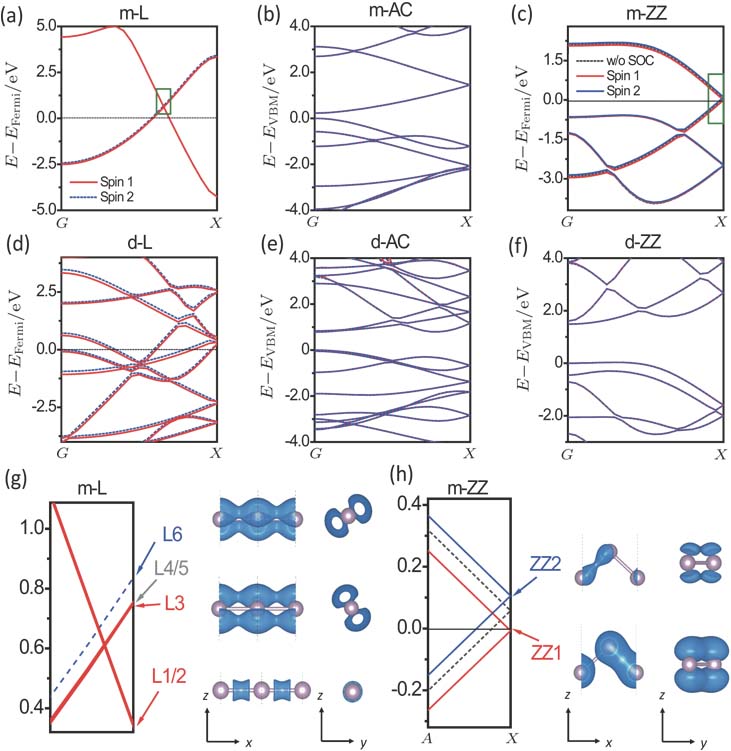
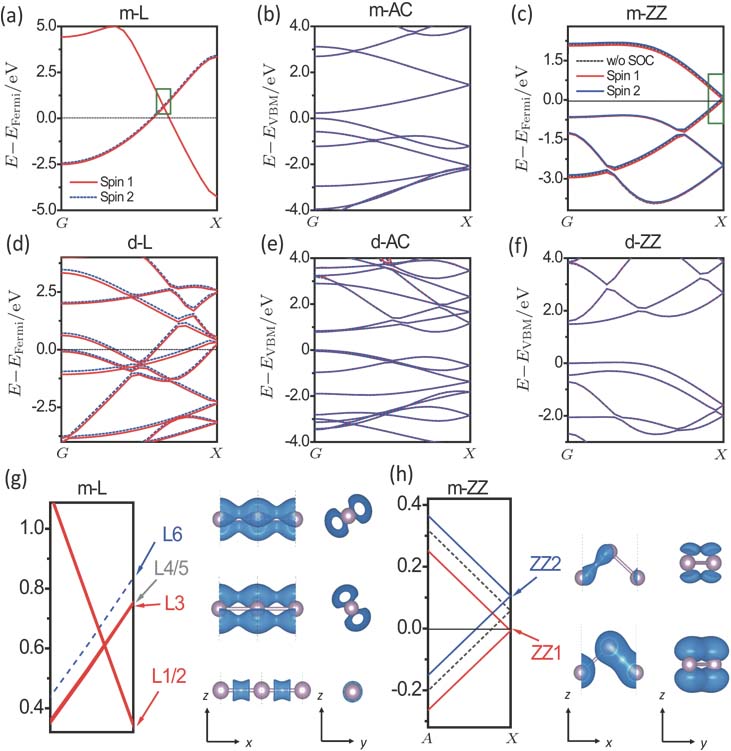 | Fig. 3. (color online) Band structures with SOC of infinite m-L (a), m-AC (b), m-ZZ (c), d-L (d), d-AC (e), and d-ZZ (f) chains. Red solid lines and blue dashed or solid lines distinguish two spin components. Black dashed lines in panel (c) indicate the bandstucture of m-ZZ without SOC. Band structures in pannels (g) and (h) are zoom-in plots of green rectangle in pannels (a) and (c), respectively. Three conduction states (L1/2, L3, L6) of m-L are marked in the left pannel of (g). Spatial structures of wavefunctions for the three marked states are shown in the right pannel of (g) using isosurface of 0.005 e·Bohr −3. One valence and one conduction states (ZZ1, ZZ2) are marked in the left pannel of (h). Spatial structures of wavefunctions for the two marked states are shown in the right pannel of (h) using isosurface of 0.003 e·bohr −3. The coordinates of A and X points are (0.4 0 0) and (0.5 0 0). |
3.4. Encapsulated finite P chains
In analog to pioneering studies on atomic chains, e.g., graphene,[5] BN,[7] or MoS2,[8] mono- or few-atom atomic chains were usually realized as finite chains encapsulated between two nanoribbons. Here, we investigated over 120 configurations of mon- and dual-atomic finite phosphorus chains encapsulated in two BP ribbons with (supported finite chains) and without BP sheets underneath (suspended finite chains). The BP sheets underneath usually stabilize the chain through the chain–sheet, mostly van der Waals, interaction.[35–37] However, even with a BP sheet, supported encapsulated linear, either for mono- or dual-atomic, chains collapse into isolated atoms, atomic clusters, or ZZ chains attaching to the BP sheet, in consistent with the stability orders found in the infinite chains. Finite ZZ chains are, again, the most stable configurations among all finite mono- and dual-atomic chains. Figures
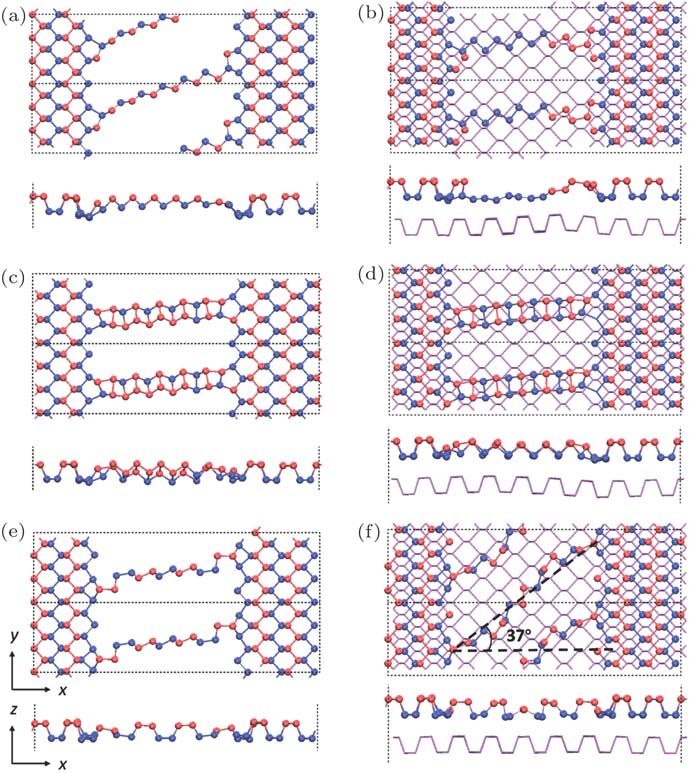 | Fig. 4. (color online) Atomic structures of the most stable configuration of suspended ((a), (c), (e)) and supported ((b), (d), (f)) finite m-ZZ ((a), (b)), d-ZZ ((c), (d)), and m-AC chains ((e), (f)). |
Finite AC chains are less stable compared with ZZ chains. However, m-AC chains can be either obtained in suspended or supported forms if n is in a proper range for AC chains, e.g., 1.32–1.40 Å. Figure
4. Conclusion and perspectives
In summary, we investigated the geometric stability and electronic properties of infinite, suspended, and supported finite mono- and dual-atomic BP chains. Regardless the width and length of the chain, the zigzag form is always the most energetic stable one and the linear chain is difficult to realize and rather fragile under compression of stretching. As the phase diagram suggested, AC chain can be reversibly transferred to ZZ chain if varying the length density of P atoms, n, from the range of AC chains (1.3–1.4 Å) to that of ZZ chains (1.5–1.6 Å). This range does not substantially change among infinite, suspended, and supported finite chains. Strong SOC effect is observed in both linear chains and the m-ZZ chain. It turns out that the infinite m-ZZ is most likely a Dirac semimetal with the Dirac point slightly off the X point. In terms of finite chains, m-ZZ is again the most stable one among those three forms. No appreciable angle dependence is found for suspended and supported finite chains except for the supported m-AC chain that it prefers the [110] direction of BP sheet, roughly 35° to 37° with respect to the normal direction of BP. This work reveals the geometric stability of various P atomic chains and shows their transitions from one form to another, especially between AC and ZZ chains. In addition, the fairly stable m-ZZ chain was predicted a Dirac semi-metal with extremely high carrier mobility. These results pave the way of extending the world of mono- or few-atom chains from graphene, BN, and TMD to BP, which may promote future research on electronic and optoelectronic properties of BP chains.
Note that during preparing this manuscript, we were aware that likely mono-atomic BP chains were fabricated and observed in a very recent transmission electronic microscope experiment.[34]
Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] | |
| [32] | |
| [33] | |
| [34] | |
| [35] | |
| [36] | |
| [37] |