

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Structures, stabilities, and magnetic properties of the FenAu (n = 1−12) clusters
[Lv Jin†,  , Zhang Jiang-Yan, Liang Rui-Rui, Wu Hai-Shun]
, Zhang Jiang-Yan, Liang Rui-Rui, Wu Hai-Shun]
, Zhang Jiang-Yan, Liang Rui-Rui, Wu Hai-Shun]
|
|


† Corresponding author. E-mail:
Project supported by the National Natural Science Foundation of China (Grant No. 21301112) and the Ph. D. Program Foundation of the Education Ministry of China (Grant No. 20131404120001).
The configurations, stabilities, electronic, and magnetic properties of FenAu (n = 1−12) clusters are investigated systematically by using the relativistic all-electron density functional theory with the generalized gradient approximation. The substitutional effects of Au in Fen+1 (n = 1, 2, 4, 5, 10−12) clusters are found in optimized structures which keep the similar frameworks with the most stable Fen+1 clusters. And the growth way for FenAu (n = 6−9) clusters is that the Au atom occupies a peripheral position of Fen cluster. The peaks appear respectively at n = 6 and 9 for FenAu clusters and at n = 5 and 10 for Fen+1 clusters based on the size dependence of second-order difference of energy, implying that these clusters possess relatively high stabilities. The analysis of atomic net charge Q indicates that the charge always transfers from Fe to Au atom which causes the Au atom to be nearly non-magnetic, and the doped Au atom has little effect on the average magnetic moment of Fe atoms in FenAu cluster. Finally, the total magnetic moment is reduced by 3 μB for each of FenAu clusters except n = 3, 11, and 12 compared with for corresponding pure Fen+1 clusters.
Nanoclusters have aroused considerable interest recently, mainly due to their novel electronic and magnetic properties which are different from those of the individual atoms and molecules or bulk matter. Of transition metal clusters, Fe clusters are the most widely studied 3d transition-metal clusters both experimentally[1–5] and theoretically.[6–14] The popularity of iron cluster is attributed to its distinctive reactivity, electronic, magnetic, and catalytic properties[15–21] as cluster size varies. Besides these properties, iron is well known for its high magnetization and an easy conversion to biocompatible oxide, which has received much attention in the biomedical areas.[22–24] At the same time, gold has been regarded as a special research material because of its unique features such as catalytic activity and bio-compatibility.[25–31] Therefore, much attention has been paid to studying the nanostructures comprised of iron and gold in both basic science research and promising application.
The bimetallic Fe/Au nanoparticles have become more popular, which is due mainly to the outstanding physical and chemical properties. Of particular interest is good bio-compatibility associated with biological applications. Naturally, based on this reason, a great number of researches have been done. Recently, Liu et al.[32] have reported that iron–gold (FeAu) alloy nanoparticles with the optical and magnetic properties present a promising prospect in biological applications. Sun et al.[33] studied that the gold-coated iron nanoclusters prevent the iron core from oxidizing, perhaps making a chance to functionalize drug delivery for the magnetic particles. Obviously, Wijaya et al.[34] performed magnetic heating experiments of Fe-doped Au nanoparticles and found that the nanoparticles exhibit notable magnetic behavior, presenting a great potential for use in hyperthermia application. Besides, experimental researches about Au–Fe alloys also have been performed in order to explore new nanomaterials in many functional material applications. The optical investigation of AuFe alloys are studied with the Fe concentration range by Gorshunov et al.[35] And they observed that the ferromagnetic AuFe clusters give rise to partial localization of conduction electrons. Via a combination of x-ray magnetic circular dichroism (XMCD) experiments and superconducting quantum interference device (SQUID) magnetometries, Wilhelm et al.[36] have demonstrated that the Fe in fcc disordered Au–Fe alloy exists in the form of the high-spin state and found the maximum value of magnetic Au moment in bcc Au–Fe alloys. Meanwhile, Crespo et al.[37] reported a crucial experiment, showing that Fe impurities weaken the ferromagnetic behavior in Au-contained nanoparticles. Moreover, theoretically, Sternik and Parlinski[38] calculated bilinear coupling constant for Fen/Aum multilayers and observed the increased magnetism of Fe atom, where there is a slight polarization of Au layers. To sum up, the extent to which size, shape, and composition of iron and gold are controlled will be a cruical factor of its flexible applications in magnetic field and biological area.
Based on the above analysis, a considerable number of theoretical researches about Fe–Au alloy clusters have been carried out. Up to now, these investigations mainly focused on the gold-rich Fe–Au alloy clusters aiming at obtaining the desired structural, magnetic, and chemical properties for potential applications. For example, Die et al.[39] have investigated single Fe doped gold clusters by using the density-functional theory and summarized the low-energy isomers tending to a two-dimensional structure. Interestingly, Zhang et al.[40] revealed doping 3d transition-metal Fe atom into gold cluster that is of planar hexagonal structure where it could stabilize the Au6 ring and promote the formation of new binary alloy clusters. The stabilities of gold clusters doped with open 3d-shell Fe atom have been investigated by Torres,[41] demonstrating that the electronic and magnetic structure depend on the dopant atom and the geometrical environment. Notably, Yang et al.[42] obtained the result that Fe@Au24 cluster still keeps the tubular Au24 structure with a slight distortion and retains the atom-like magnetism of 4 μB, which might be used as new nanomaterials with tunable magnetic properties in the near future. Later, Wang et al.[43] found that 
Motivated by the above research background, we carry out the present study on the gold-doped iron clusters FenAu (n = 1−12) in order to systematically explore their geometric structures, relative stabilities, electronic and magnetic properties. We attempt to clarify the following points: (i) how the Au doping influences the geometrical structures of the Fen cluster, (ii) what the size-dependent growth behavior for FenAu is, (iii) whether some novel properties could be found when one Au atom is doped into iron clusters, (iv) how much is the degree to which the magnetic behavior is changed through incorporating the Au atom and what is the magnetic physics origin responsible for this change. It is expected that in the future, our present work can provide some forceful theoretical guidance in the Stern–Gerlach (SG) experiment, the development of good catalysts for synthesis and the biological and biomedical applications.
All calculations are performed at the DFT level with the DMol3 package in the Materials Studio of Accelrys Inc.[48,49] The electron density functional is treated with the generalized gradient approximation (GGA) corrected exchange potential of Perdew, Burke, and Ernzerhof (PBE).[50] The double numerical basis set augmented with d-polarization and p-polarization functions (DNP) is utilized. Relativistic calculations are performed with scalar relativistic corrections to valence orbitals relevant to atomic bonding properties via a local pseudopotential (VPSR).[51] For the numerical integration, a fine quality mesh size is used, and the real space cutoff of the atomic orbital is set to be 5.5 Å. The convergence criteria for structure optimization and energy calculations are set to be fine with the tolerance for electron density and total energy convergence in self-consistent field (SCF), energy, gradient, and displacement of 1 × 10−6 a.u. (The unit a.u. is short for atomic unit), 1.0 × 10−5 a.u., 0.002 a.u., and 0.005 Å, respectively. In the geometry optimization procedure, we consider a number of initial structures including linear chains, planar and three-dimensional structures in this work to maximize our chance to find the ground state configurations of the alloy clusters. First, we identify the low-lying structures of pure Fen clusters based on earlier theoretical work on iron[6–14] and other TM clusters,[52–,55] and choose these low-lying structures as various reasonable initial structures. Second, on the basis of these initial structures, we substitute the Fe atom with the Au atom at different positions of Fen+1 cluster, and we place the Au atom on each possible site of Fen cluster, and also test some structures through referring to ConMn/ConV,[56,57] ScnAl,[58] FenMn,[59] FenCr,[60] ConAu,[61,62] ConFe,[63] FenPt,[64] and YnAl[65] clusters. In this way, we consider more than 400 candidates altogether. Moreover, without any symmetry constrains, we relax the geometric structure to find out the true ground state. Third, for each geometry structure, the magnetic moment is first allowed to optimize automatically into the favored state (Sz) in DMol3, then, we consider the neighboring spin states (Sz±2) or longer-range spin states and optimize them by fixing the spin state. The calculations are implemented until the minimum energy is reached. Finally, subsequent frequency calculations are performed to confirm the optimized geometry corresponding to the minimum value. In addition, for confirming that our obtained low-lying isomers are the most stable isomers in the FenAu clusters, a global search of Fe7Au cluster based on a particle swarm optimization algorithm within an evolutionary scheme is carried out by using the CALYPSO package.[66] As a result, the most stable structure and metastable state obtained from CALYPSO are consistent with those from our methods mentioned above. The other low-lying isomers are also similar, but only in the order energy there exist some differences. This indicates that the most stable and low-lying isomers our confirmed are indeed in ground state and they are the most stable isomers. Based on the above steps, the net charge and the magnetic moments are evaluated via the Mulliken population analysis.
In order to test the validity of the computational method for the description of FenAu clusters, we first optimize Fe2 and Au2 dimers. All these results are in good agreement with the reported theoretical and experimental results.[1–3,8,13,14,67–71] As listed in Table
| Table 1. Calculated values of bond length d (Å), average binding energy Eb (eV), average magnetic moment ū (μB), vibrational frequency ω (cm−1), and ionization potential IP (eV) of the Fe2 and Au2 clusters. . |
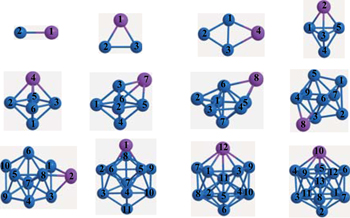
Using the computational scheme described in Section 2, we have explored a number of low lying isomers and determined the lowest energy structures of FenAu clusters up to n = 12. The obtained ground state structures which include these isomers within 0.4 eV of the most stable structure are shown in Fig.
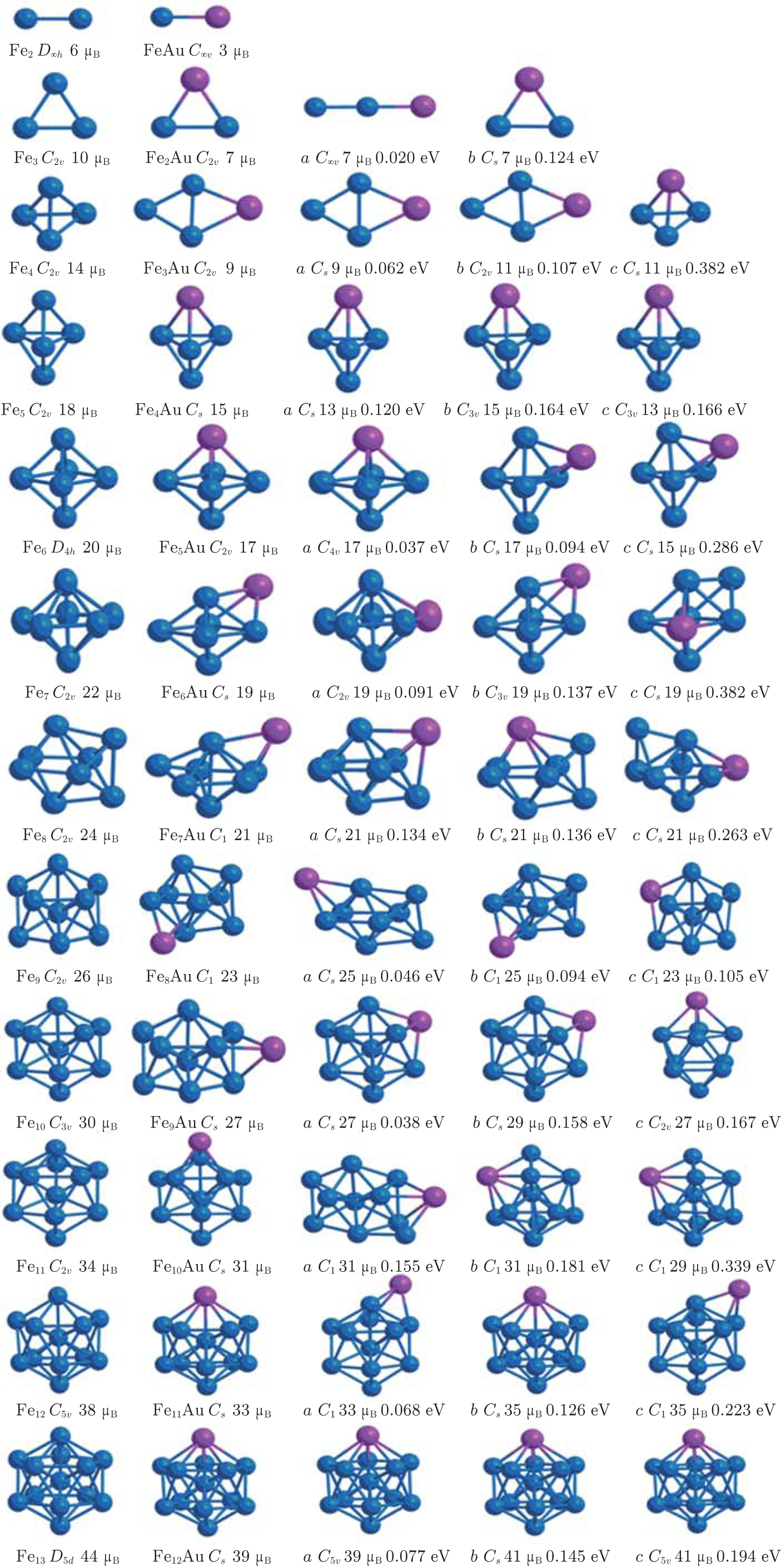 | Fig. 1. Lowest-energy structures and low-lying isomers with different relative energies (in unit eV) of FenAu (n = 1−12) clusters. The ground state geometries of the corresponding bare Fen+1 clusters are also given on the left. The blue and pink balls represent Fe and Au, respectively. Distances are given in unit Å. |
| Table 2. Values of average binding energy (Eb), average bond length (d), average magnetic moment at Fe atom (ūatom), HOMO–LUMO (highest occupied molecular orbital–lowest unoccupied molecular orbital) gaps in the lowest energy structures of FenAu clusters. Corresponding values for Fen+1 cluster are given in the brackets. . |
FeAu is C∞v symmetry dimer, whose bond length and average binding energy are 2.398 Å and 1.552 eV/atom, respectively. These results lie between Fe2 (2.020 Å, 1.621 eV/atom) and Au2 (2.488 Å, 1.189 eV/atom). Compared with it of pure Fe2 (6 μB), the magnetic moment of FeAu dimer is reduced by 3 μB. Our computed values are in good agreement with the values (2.470 Å and 3 μB) obtained in previous paper.[39]
Energetically, the most favorable structure of the Fe2Au cluster with a total magnetic moment of 7 μB is in the form of an isoscele triangle (C2v), whose Fe–Fe distance is 2.073 Å. This may be seen as replacing an Fe atom with an Au atom. Our results about Fe2Au are proved by Sun et al.,[33] whose results show the Fe–Fe distance of 2.020 Å and the same magnetism (7 μB). Its metastable state shares the linear chain structure, and the third stable state (9 μB) is similar to the ground state. They are energetically less stable by 0.020 eV and 0.124 eV, respectively.
A planar structure (C2v) is the most stable Fe3Au cluster with 9 μB of total magnetic moment. The second energetically degenerate structure (9 μB), which is of a turnup rhombus (four atoms are not on the same plane) structure (Cs), is energetically less favorable than the most stable structure in energy by 0.062 eV. The third lowest-energy structure is similar to the most stable structure, but they differ by 11 μB of total magnetic moment. A distorted tetrahedronal configuration with Cs symmetry is the fourth stable isomer. The energy is 0.382 eV less stable in energy compared the lowest energy structure, which means that there is the turning from the planar (2D) structure to the three-dimensional (3D) structure.
In the case of Fe5, we find the C2v triangular bipyramid structure to have the lowest energy. For Fe4Au, the overall structures are totally retained, but they are slightly distorted triangular bipyramid with the Au atom at the vertex. The most stable isomer is of Cs symmetry and it has a total magnetic moment of 15 μB, which is reduced by 3 μB in comparison with the pure Fe5 (18 μB). By contrast, the second stable structure (Cs) with a total magnetic moment of 13 μB is 0.120 eV evidently higher in energy. The other two stable isomers (C3v) have total magnetic moments of 15 μB and 13 μB, and 0.164 eV and 0.166 eV above the lowest-energy structure, respectively.
The C2v configuration of Fe5Au with the magnetism (17 μB) is found to be the most stable isomer, which may be viewed as a deviation from substitution of a Fe atom by an Au atom in the octahedron Fe6 (D4h) structure. The other distorted octahedron geometrical isomer with C4v symmetry is considered to be the second stable structure at a slightly higher energy (0.037 eV). Differently, the following two stable isomers prefer to share a capped triangle bipyramid with Au atom being at the cap position, which are located at 0.094 eV and 0.286 eV in energy above the most stable conformation.
For the Fe6Au clusters, we consider a lot of initial geometries including the bicapped triangular bipyramid, capped octahedron, pentagonal bipyamid, bicapped square pyramid, etc. At last, an Au-capped octahedron structure with Cs symmetry is found to have the lowest energy. However, the metastable state is a distorted pentagonal bipyramid (C2v) with the impurity Au atom in the middle pentagonal ring plane, and is found to be only 0.091 eV less stable than the ground state. The third state also favors a capped octahedron (C3v) and is energetically less favorable than the most stable structure by 0.137 eV. The fourth state is still a capped octahedron (Cs) with the Au atom at the square site, and is 0.382 eV higher in total energy. Moreover, we notice that these four stable structures of Fe6Au have the equal total magnetic moment (19 μB).
Energetically the most favorable structure for Fe7Au (21 μB) is an Au-capped distorted pentagonal bipyramid with C1 symmetry. The metastable state with a classic bicapped octahedron structure is 0.134 eV in energy higher than the lowest-energy structure. As seen in Fig.
In the case of n = 8, the most stable Fe8Au (23 μB) with C1 symmetry is capped one Au atom on the Fe8 ground-state geometry. The second stable isomer and the third stable isomer have similar geometric configurations with the lowest-energy structure but Au atoms occupy the different capped positions, whose total energies are only 0.046 eV and 0.094 eV above the lowest-energy isomer. The last stable isomer (C1) with a total magnetic moment of 23 μB can be viewed as the Fe atom in the ground state structure of Fe9 replaced by one Au atom.
For the case of Fe9Au, the lowest-energy structure with Cs symmetry prefers one capping Au atom on the surface of the ground-state Fe9 cluster. As shown in Fig.
In terms of Fe10Au (Cs), the most stable structure is similar to that of the Fe11 cluster which is a tetra-capped pentagonal bipyramid with the Au atom at the vertex position. It has a total magnetic moment 31 μB, and is 0.155 eV in energy lower than that of the metastable state. As seen in Fig.
Just like the Fe12 cluster, the lowest-energy structure of Fe11Au cluster takes an icosahedral growth pattern where Au atom substitutes the Fe site at the apex with a total magnetic moment of 33 μB as shown in Fig.
With regard to Fe12Au cluster, we consider the icosahedral, hexagonal close packed (hcp), rhombic dodecahedral (bcc) and cuboctahedral (fcc) structures as initial geometries. Overall, the most stable configurations we have finally obtained can be looked upon as a substitutional Au impurity in the pure Fe13 cluster, in which Au atom lies at the one vertex site of the icosahedral structure. As shown in Fig.
The overall evolutionary trends for the FenAu series show that for n = 1−2, 4−5, 10−12, the Au doped geometries are similar to those of Fen+1 clusters, where the Au atom occupies a substitutional surface site accompanied with a slight distortion in the host cluster. And the way of growing FenAu (n = 6−9) clusters is that the Au atom occupies a peripheral position of Fen cluster. This phenomenon can be attributed to the two aspects: on the one hand, Au atom prefers to occupy peripheral location, which can be explained by the cohesive energy (CE) and the surface energy (SE) of the metal atom. To minimize the total energy, the atom with the smaller surface energy and cohesive energy tends to occupy the surface, while the atom with a higher surface energy and cohesive energy favors the interior[72–74] The surface energy and average cohesive energy of the bulk Au are 1.506 J·m−2 and 3.81 eV/atom, which are smaller than those of the bulk Fe 2.417 J·m−2 and 4.28 eV/atom respectively.[75,76] On the other hand, the atomic size effect also accounts for this. The smaller atoms tend to occupy the more sterically confined core, especially in icosahedral clusters, exhibiting the compression effects finally forming a stable cluster.[72] The atomic radius of Au is larger than that of Fe atom.[75] Thus, the Fe atoms prefer to occupy central position, whereas the Au atom prefers to be located at the site of surface. Similar phenomena and explanations also appeared in previous studies.[61,76–79]
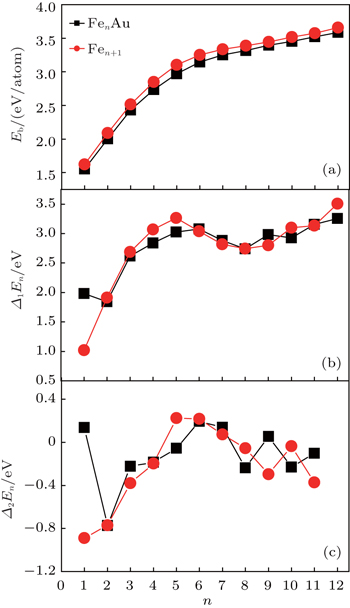
In this subsection, we will investigate the size-dependent physical properties of these clusters. The variation of average binding energy Eb(n) with cluster size is shown in Fig.
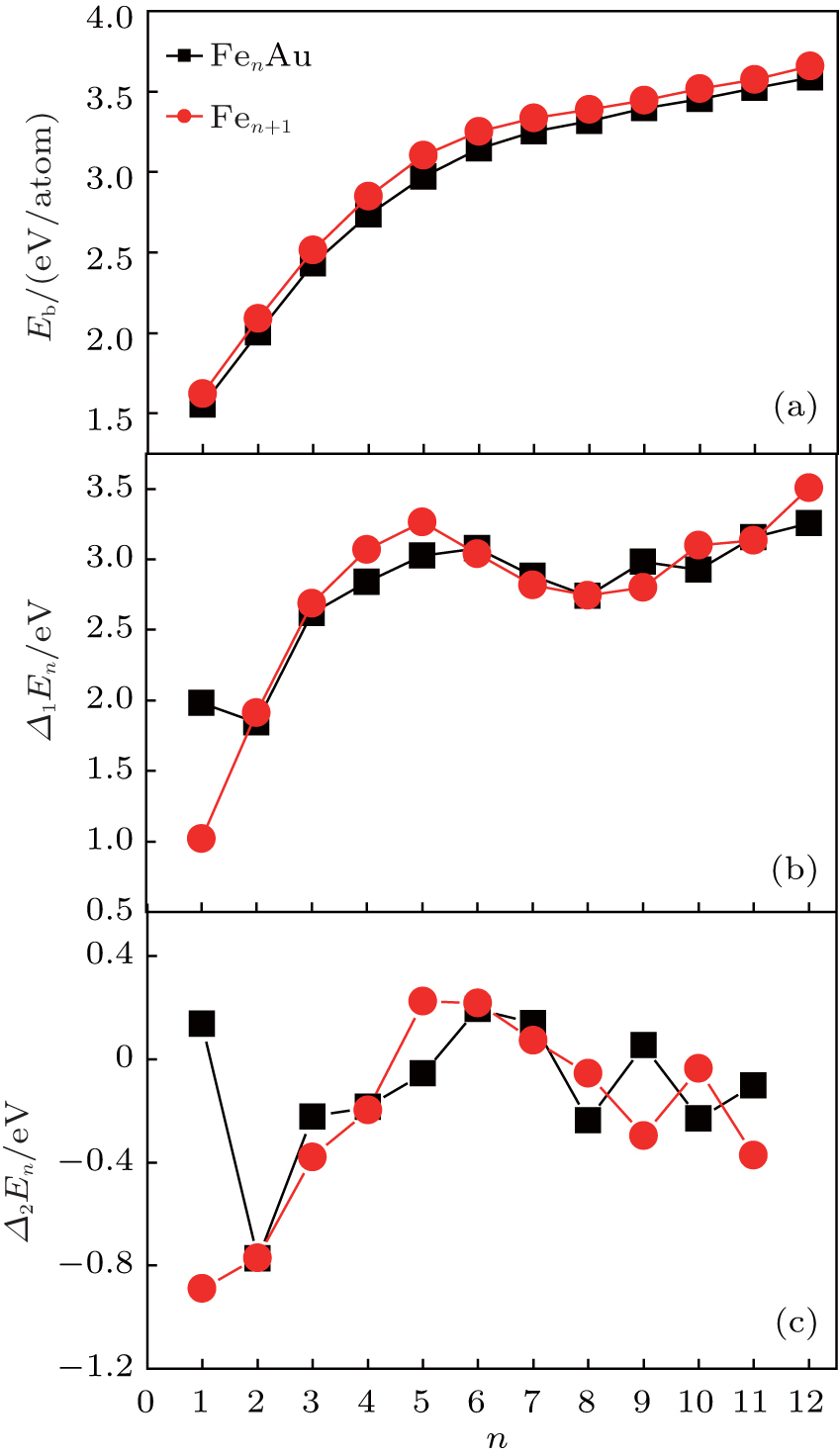 | Fig. 2. Size dependences of averaged atomic binding energies (a), fragmentation energies (b), and second-order difference energies (c) for the lowest energy structures of FenAu and Fen+1 (n = 1−12) clusters. |
In cluster physics, the fragmentation energies and second-order difference of energies are sensitive indicators of the relative stability. Thus, the size dependence of the fragmentation energies and second-order difference of energies for FenAu (n = 1−12) clusters are further investigated. The fragmentation energy Δ1E(n) and the second-order difference energy Δ2E (n) are calculated using the following formulas:
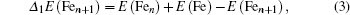
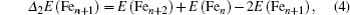
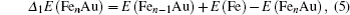
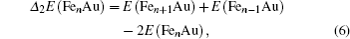
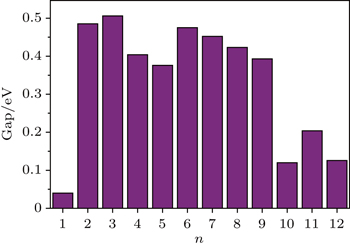
In addition, the HOMO–LUMO gap is a characteristic quantity of electronic structure in clusters and is commonly used to measure the ability for clusters to undergo activated chemical reactions with small molecules. A big energy gap usually corresponds to a high chemical stability. As shown in Fig.
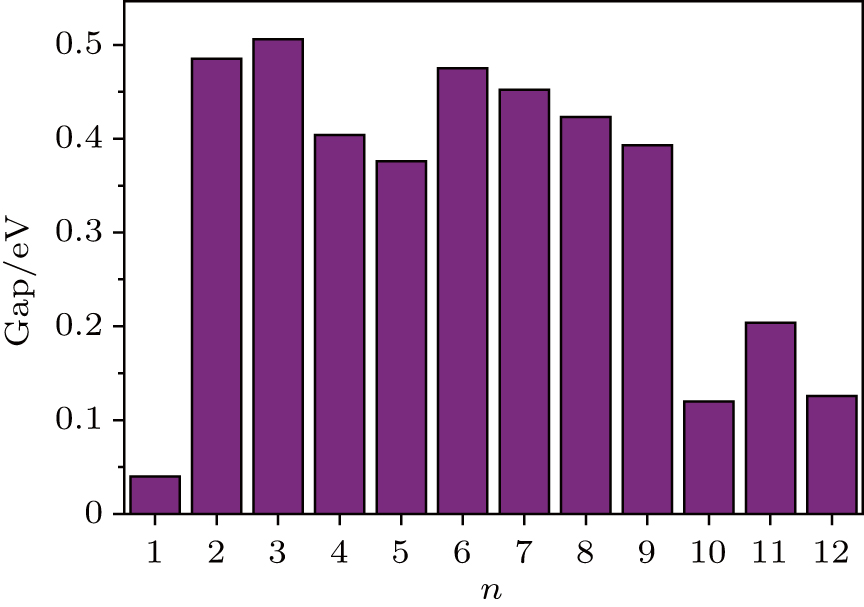 | Fig. 3. HOMO–LUMO gaps of the most stable structures of FenAu clusters as function of cluster size. |
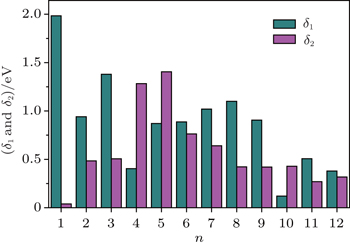
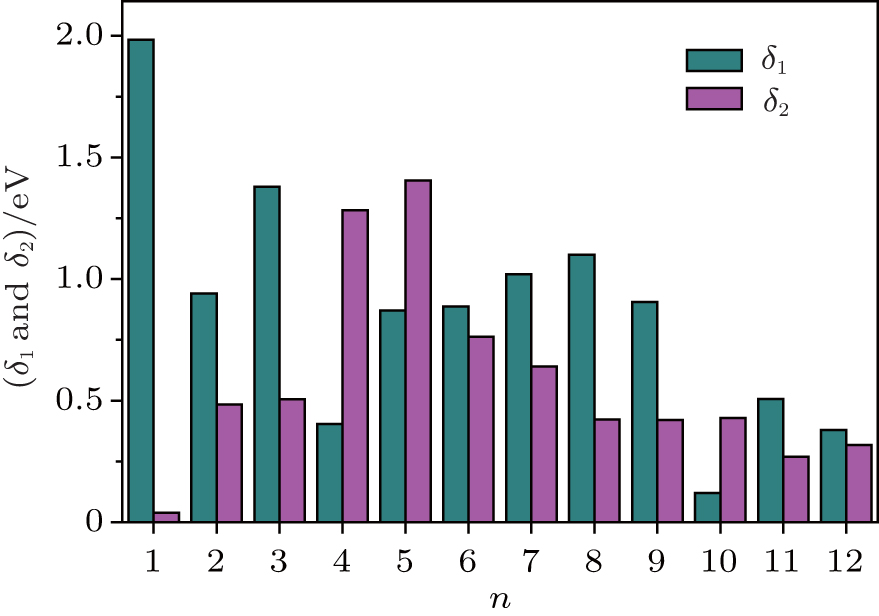
The magnetic stability of these clusters can be verified by calculating the spin gaps as a function of the cluster size. The spin gaps for a magnetic cluster are defined as
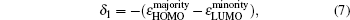
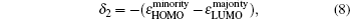
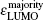

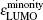
 | Fig. 4. Size dependences of the spin gaps of FenAu (n = 1−12) clusters. |
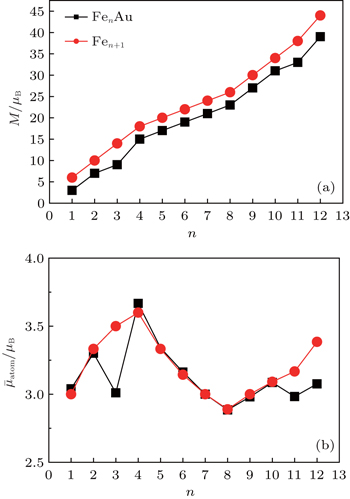
The total magnetic moments of the ground-state FenAu (n = 1−12) clusters are calculated and the results are presented in Fig.
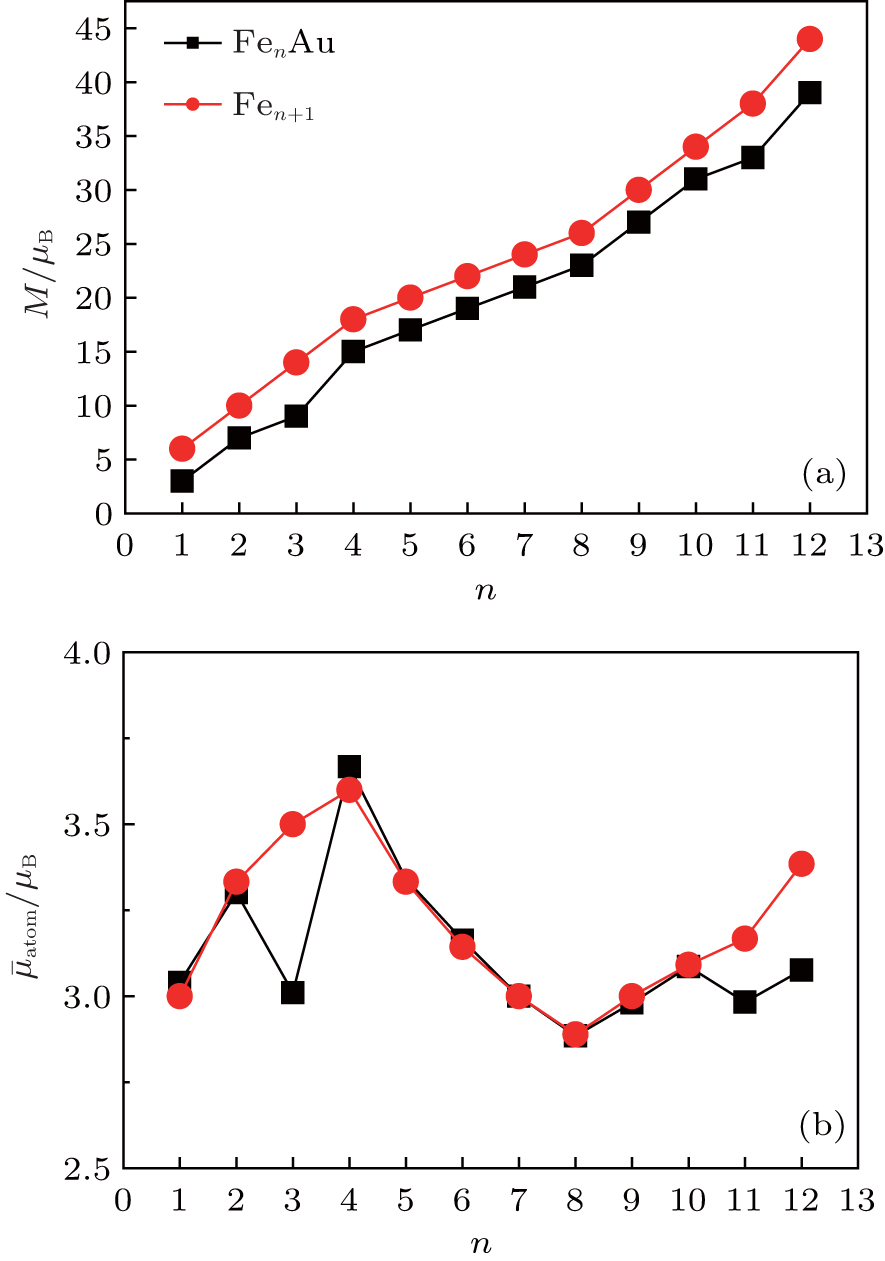 | Fig. 5. Size dependences of the magnetic moments (a) and the average magnetic moments of Fe atom (b) for FenAu and Fen+1 (n = 1−12) clusters. |
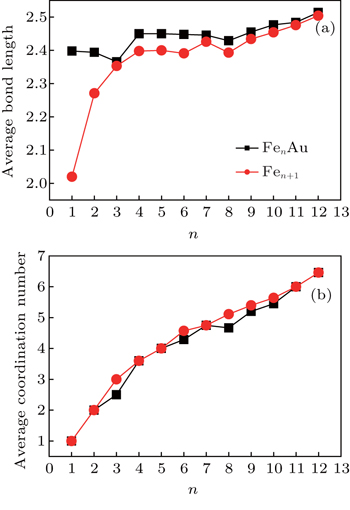
As shown in Fig.
 | Fig. 6. Variations of average bond length (a) and average coordination number (b) with the cluster size for FenAu and Fen+1 (n = 1−12) clusters. |
The reason why the Fe atomic average magnetic moments of n = 3, 11, and 12 alloy clusters obviously decrease compared with that of pure Fe clusters is explained in detail. It can be seen from Fig.
For the optimized geometry structure, the total magnetic moments of FenAu clusters, the local moments of Fe and Au atoms, the atomic net charges and the local moments of 3d, 4s, and 4p states for Fe and Au atoms are calculated and summarized in Table
| Table 3. Atom labels and values of atomic net charge Q (e), local magnetic moment μatom (μB) of the atom, Mulliken charge (e), and local magnetic moment of s, p, and d states μL (μB) for the lowest energy structures of FenAu (n = 1−12) clusters. . |
 | Fig. 7. Atom labels of the ground state of FenAu (n = 1−12) clusters. |
To gain an insight into the magnetism of the FenAu cluster, we also explore the spin densities of the alloy clusters. In Fig.
 | Fig. 8. spin densities of FenAu and Fen+1 (n = 1−12) clusters. The density isosurfaces are drawn at 0.5 a.u. |
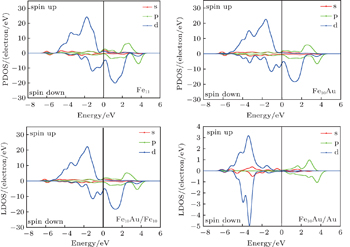
In order to explore the magnetic origin further, In Fig.
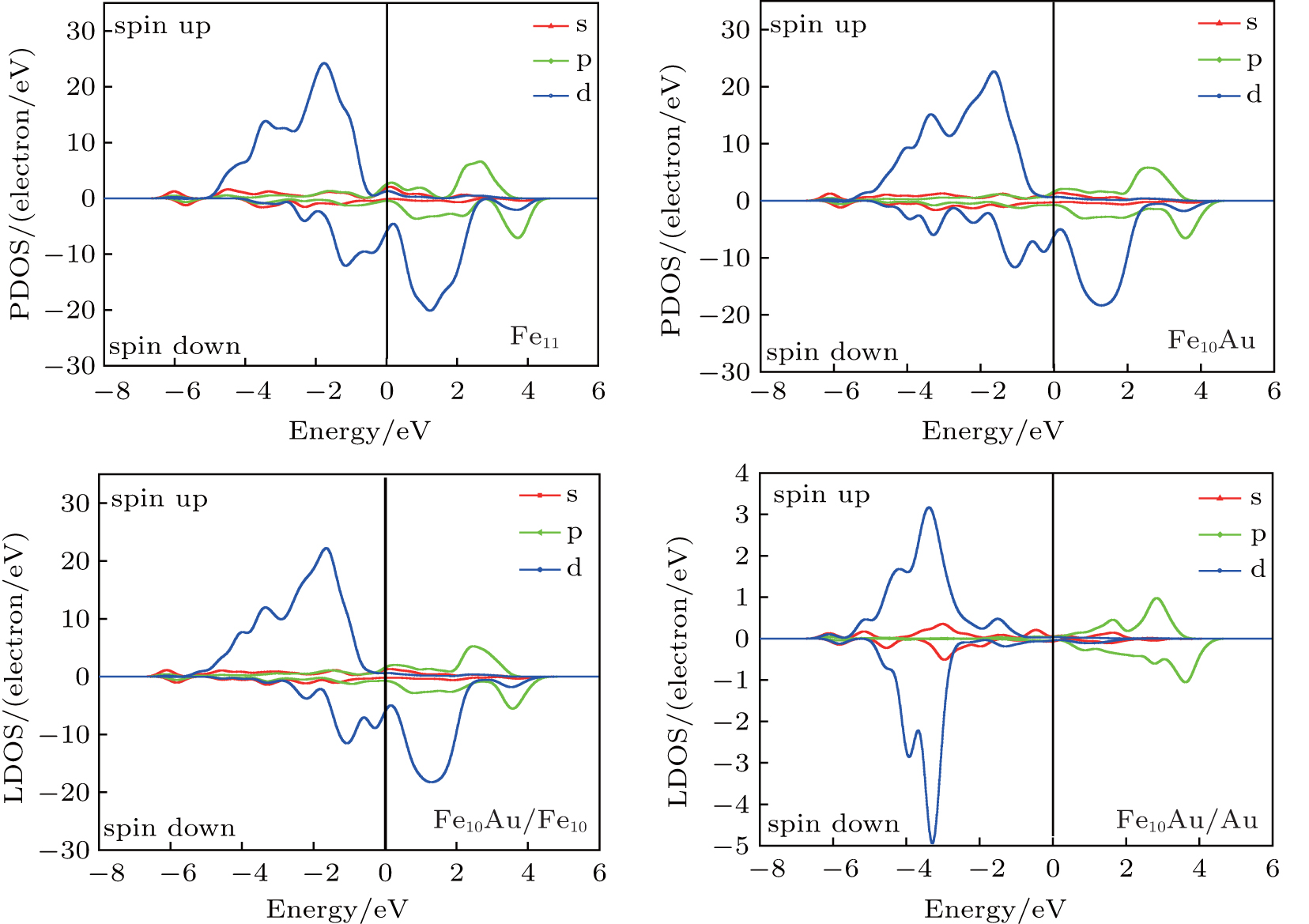 | Fig. 9. PDOSs and LDOSs of Fe10Au and Fe11 clusters. |
In this work, we perform DFT–GGA computations of FenAu (n = 1−12) alloy clusters. The size-dependent structural, electronic, magnetic properties are investigated. The main conclusions are summarized as follows.
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 | |
| 22 | |
| 23 | |
| 24 | |
| 25 | |
| 26 | |
| 27 | |
| 28 | |
| 29 | |
| 30 | |
| 31 | |
| 32 | |
| 33 | |
| 34 | |
| 35 | |
| 36 | |
| 37 | |
| 38 | |
| 39 | |
| 40 | |
| 41 | |
| 42 | |
| 43 | |
| 44 | |
| 45 | |
| 46 | |
| 47 | |
| 48 | |
| 49 | |
| 50 | |
| 51 | |
| 52 | |
| 53 | |
| 54 | |
| 55 | |
| 56 | |
| 57 | |
| 58 | |
| 59 | |
| 60 | |
| 61 | |
| 62 | |
| 63 | |
| 64 | |
| 65 | |
| 66 | |
| 67 | |
| 68 | |
| 69 | |
| 70 | |
| 71 | |
| 72 | |
| 73 | |
| 74 | |
| 75 | |
| 76 | |
| 77 | |
| 78 | |
| 79 | |
| 80 | |
| 81 | |
| 82 | |
| 83 | |
| 84 | |
| 85 | |
| 86 |