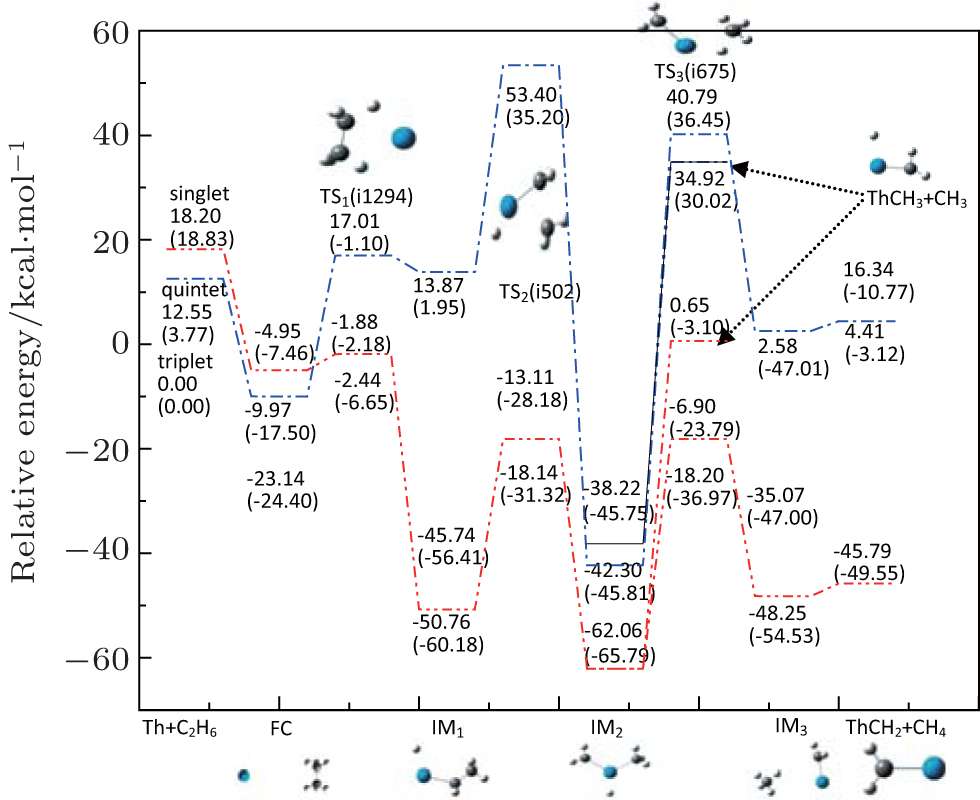
Density function theoretical study on the complex involved in Th atom-activated C–C bond in C2H6
Wang Qing-Qing1, Li Peng2, Gao Tao1, †,  , Wang Hong-Yan3, Ao Bing-Yun4
, Wang Hong-Yan3, Ao Bing-Yun4
, Wang Hong-Yan3, Ao Bing-Yun4 Geometric structures and potential energy profiles for the reaction of Th + C2H6, computed at the B3LYP/SDD and PW91/SDD (in parentheses) levels of theory.