

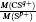
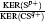
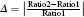
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Coulomb explosion of CS2 molecule under an intense femtosecond laser field
[Wang Xiao1, Zhang Jian1, Zhang Shi-An1, 2, †,  , Sun Zhen-Rong1]
, Sun Zhen-Rong1]
, Sun Zhen-Rong1]
|
|


† Corresponding author. E-mail:
Project supported by the National Natural Science Foundation of China (Grant Nos. 51132004 and 11474096), and the Science and Technology Commission of Shanghai Municipality, China (Grant No. 14JC1401500). We acknowledge the support of the NYU-ECNU Institute of Physics at NYU Shanghai, China.
We experimentally demonstrate the Coulomb explosion process of CS2 molecule under a near-infrared (800 nm) intense femtosecond laser field by a DC-sliced ion imaging technique. We obtain the DC-sliced images of these fragment ions S+, S2+, CS+, and CS2+ by breaking one C–S bond, and assign their Coulomb explosion channels by considering their kinetic energy release and angular distribution. We also numerically simulate the dissociation dynamics of parent ions 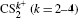
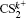
With the dramatic development of ultrashort laser technique in the past few decades, the ultrashort intense laser field has brought out some unprecedented physical phenomena, such as multi-photon ionization (MPI),[1–3] enhanced ionization (EI) or charge-resonance enhanced ionization (CERI),[4–9] above-threshold ionization (ATI),[10] dissociative ionization,[11–14] Coulomb explosion (CE),[15–23] and so on. As is well known, when the laser intensity is up to 1014 W/cm2, the laser field magnitude is comparable to the Coulomb field generated by an atomic nucleus, the molecular geometry structure can be modified, and thus the molecular chemical bond will be broken by the strong Coulomb repulsive force. Several experimental results indicated that the chemical bond will be stretched from the equilibrium nuclear distance (Re) in the neutral molecule to a critical distance (Rc), and the CE process occurs at this critical distance.[6,23–28] Schmidt et al.[29] explained this phenomenon on the basis of a laser-induced stabilization, and found that the bond elongation is mainly determined by the molecule itself. Based on this viewpoint, Corkum et al.[4] proposed a theoretical model of laser-induced electron localization enhanced ionization, which concluded that the ionization of the parent ion will be dramatically enhanced around the critical distance Rc since the electron can directly tunnel through the narrow internal barrier to the continuum. Similarly, Bandrauk et al.[5,6] presented a charge resonance enhanced ionization model, and showed that the enhanced ionization probability at the critical distance Rc is attributed to the transitions between a pair of charge-resonant states that are strongly coupled to the laser field.
As a prototypical linear triatomic molecule, the photoionization and photodissociation of CO2 and CS2 molecules under the intense femtosecond laser fields have been extensively studied. As a representative study of the CO2 molecule, Wu et al.[30,31] studied the three-body fragmentation dynamics of CO2 molecule in intense laser fields by using a triple ion coincidence technique, and showed that the geometric structure of 

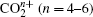
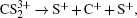
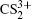
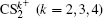
Details of our home-built DC-sliced ion imaging system have been described in our earlier publication,[27] and only a brief description is given here. The CS2 sample (99.9% purity) carried by 1 atm helium gas is expanded adiabatically into the source chamber through a pulsed valve with the repetition rate of 100 Hz, and then skimmed into the high-vacuum main chamber with a base pressure of ∼ 10−8 mbar. The multi-stage ion lens in the main chamber has a similar configuration described by Suits et al.,[34] and the optimized voltages applied on the electrodes are URepeller = 2000 V, U1 = 1760 V, U2 = 1660 V, and U3 = 0 V. The super-sonic molecular beam interacts with the intense femtosecond laser field in the main chamber, and the fragment ions are first accelerated by a multi-lens velocity mapping apparatus and then fly freely to a dual micro-channel plate (MCP) coupled with a P47 phosphor screen. The sliced images of the fragment ions are obtained with an intensified charge-coupled device (ICCD) camera, and the time-of-flight (TOF) mass spectra are achieved by a photomultiplier tube (PMT) connected to a digital oscilloscope. All the timing sequence control is implemented by a digital delay pulse generator (DG535 Stanford Research System).
Figure 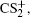

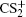
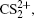
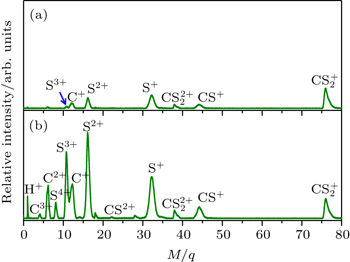
 | Fig. 1. The TOF mass spectra of CS2 molecule irradiated by 800 nm femtosecond laser pulses with a laser intensity of 4.9 × 1013 (a) and 1.3 × 1014 W/cm2 (b). |
When the polyatomic molecules are subjected to an intense femtosecond laser field, the fragment ions can be produced from different photoionization and photodissociation channels. With the help of DC-sliced image technique, we can clearly discriminate these different dissociative ionization channels of the fragment ions with same mass-to-charge ratio. Figure


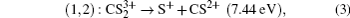
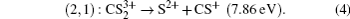

Table 
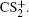
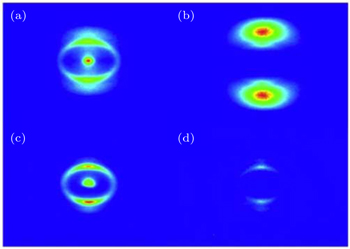
 | Fig. 2. The pseudo-color DC sliced images of fragment ions S+ (a), S2+ (b), CS+ (c), and CS2+ (d) with the laser intensity of 1.5 × 1014 W/cm2. |
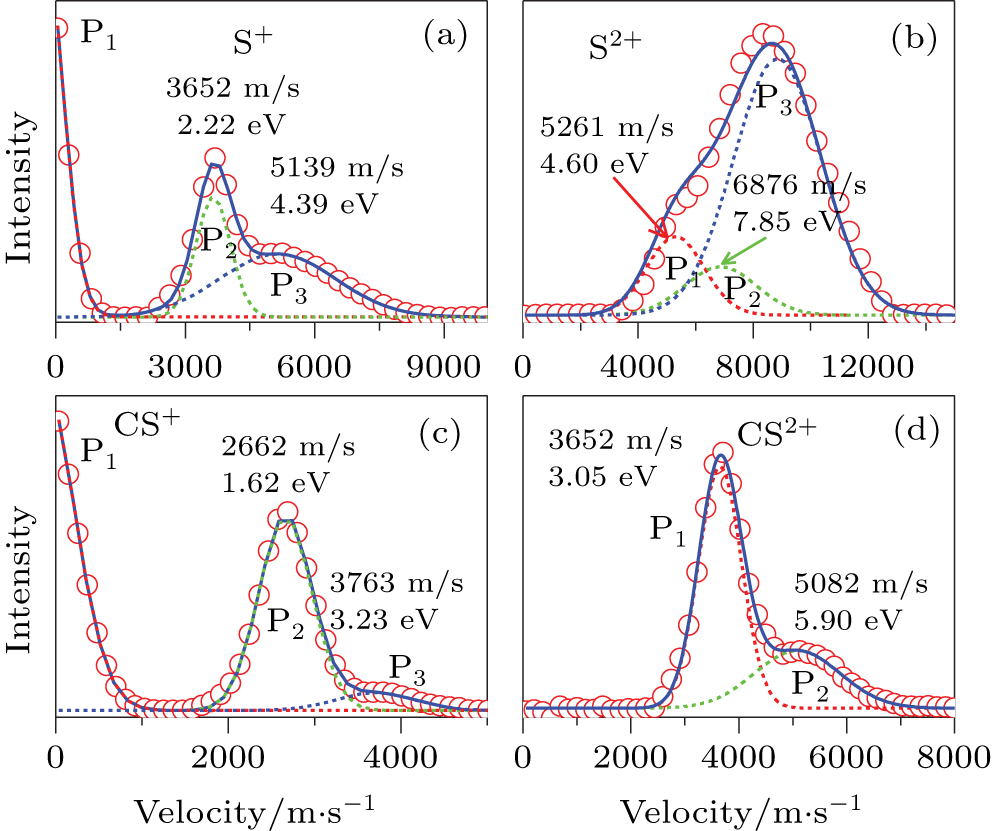 | Fig. 3. The velocity distributions of fragment ions S+ (a), S2+ (b), CS+ (c), and CS2+ (d). The red circles are the experimental data, and the blue solid lines are the simulated results by the multiple Gaussian functions. |
| Table 1. Mass ratio |
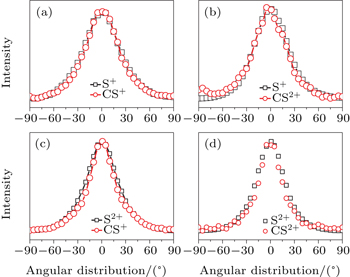
In addition to the KER distribution, the angular distribution of fragment ions also plays an important role in assigning the dissociation channels. The two fragment ions from the same dissociation channel should have the similar angular distribution. Figure
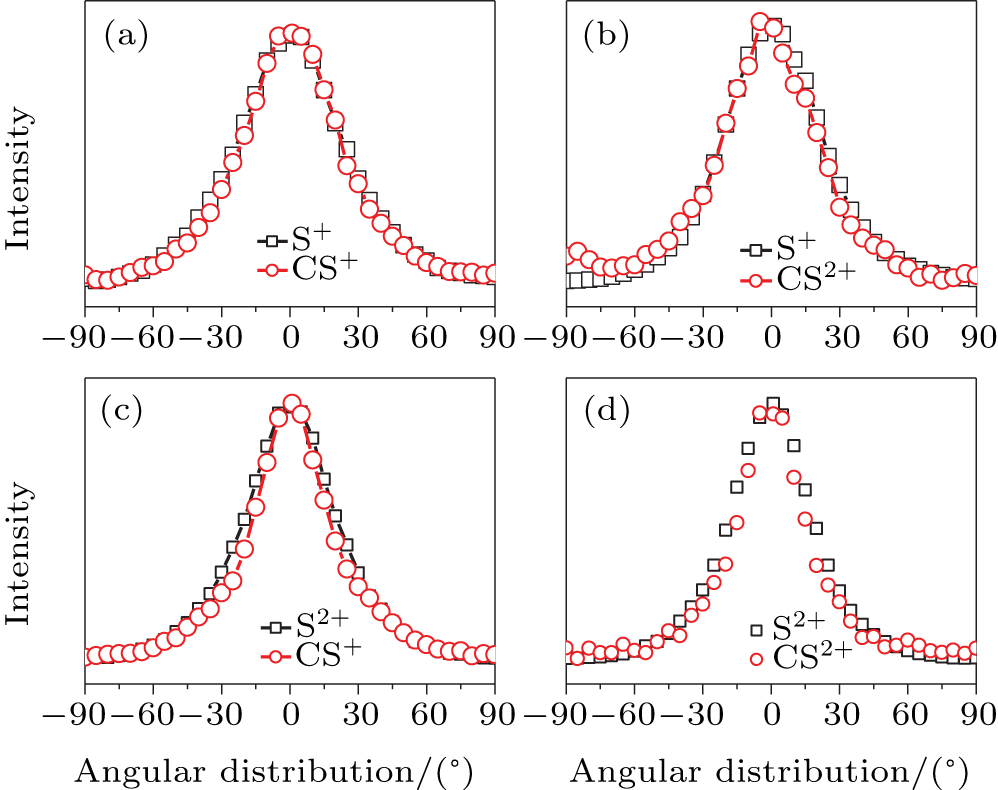 | Fig. 4. The angular distributions of the two fragment ions that correspond to the four CE channels (1, 1) (a), (1,2) (b), (2,1) (c), and (2,2) (d). |
It has been proved that the total KER Etotal from the CE process and the critical distance Rc should satisfy the following relationship:[23,26]
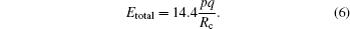
To better understand this energy conversion process, we use a Coulomb potential approximation to theoretically simulate the dissociation process of parent ions, and here the doubly charged parent ion 
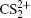
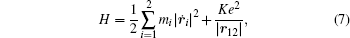
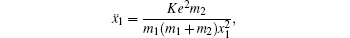
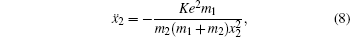

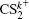
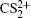

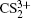
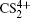
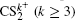
 | Fig. 5. The time evolution of Coulomb energy (red dotted lines) and KER (black solid lines) for the four CE channels 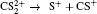  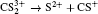  |
| Table 2. Time requirement for different percentages of Coulomb energy to KERs in the four CE channels. . |
In summary, the CE process of CS2 molecule under the near-infrared (800 nm) intense femtosecond laser field has been experimentally investigated with a DC-sliced ion imaging technique. The CE channels of fragment ions S+, S2+, CS+, and CS2+ by breaking one C–S bond were confirmed by calculating their corresponding velocity distribution and angular distribution. The Coulomb explosion process of highly charged parent ions 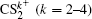
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 | |
| 22 | |
| 23 | |
| 24 | |
| 25 | |
| 26 | |
| 27 | |
| 28 | |
| 29 | |
| 30 | |
| 31 | |
| 32 | |
| 33 | |
| 34 | |
| 35 | |
| 36 | |
| 37 | |
| 38 | |
| 39 | |
| 40 | |
| 41 |