



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Accurate double many-body expansion potential energy surface of HS2A2A′) by scaling the external correlation
[Zhang Lu-Lu, Song Yu-Zhi†,  , Gao Shou-Bao, Zhang Yuan, Meng Qing-Tian‡, ]
, Gao Shou-Bao, Zhang Yuan, Meng Qing-Tian‡, ]
, Gao Shou-Bao, Zhang Yuan, Meng Qing-Tian‡, ]
|
|


† Corresponding author. E-mail:
‡ Corresponding author. E-mail:
Project supported by the National Natural Science Foundation of China (Grant No. 11304185), the Taishan Scholar Project of Shandong Province, China, the Shandong Provincial Natural Science Foundation, China (Grant No. ZR2014AM022), the Shandong Province Higher Educational Science and Technology Program, China (Grant No. J15LJ03), the China Postdoctoral Science Foundation (Grant No. 2014M561957), and the Post-doctoral Innovation Project of Shandong Province, China (Grant No. 201402013).
A globally accurate single-sheeted double many-body expansion potential energy surface is reported for the first excited state of HS2 by fitting the accurate ab initio energies, which are calculated at the multireference configuration interaction level with the aug-cc-pVQZ basis set. By using the double many-body expansion-scaled external correlation method, such calculated ab initio energies are then slightly corrected by scaling their dynamical correlation. A grid of 2767 ab initio energies is used in the least-square fitting procedure with the total root-mean square deviation being 1.406 kcal·mol−1. The topographical features of the HS2(A2A′) global potential energy surface are examined in detail. The attributes of the stationary points are presented and compared with the corresponding ab initio results as well as experimental and other theoretical data, showing good agreement. The resulting potential energy surface of HS2(A2A′) can be used as a building block for constructing the global potential energy surfaces of larger S/H molecular systems and recommended for dynamic studies on the title molecular system.
Sulfur-containing species have received growing attention due to their important role played in the air pollution and acid rain.[1–7] Among various sulfur-containing compounds, the HS2 radical is one of the most important species, which is responsible for the oxidation of reduced forms of sulfur, the reaction in combustion[8–10] and biochemistry.[11,12] Therefore, extensive studies on the HS2 radical have been carried out both theoretically and experimentally for decades.
Among a vast array of theoretical studies, Sannigrahi et al.[13] performed CI calculations and provided both structural and vibrational information of the electronic ground and excited states of HS2. Zhou et al.[14] carried out investigations on the geometries and spectroscopic parameters of the XS2 (X = H, F, Al, and Br) radicals using the split-valence plus polarization function and larger basis sets at the self-consistent field (SCF) and the second order Møller–Plesset (UMP2). By employing the CCSD(T) theory, the equilibrium structure and vibrational frequencies were reported by Owens et al.[15] for the X2H hydrides (X = Al, Si, P, and S) at their global minima. Subsequently, more accurate structural and thermodynamic properties of the HS2 radical were obtained by Denis[16] using the CCSD(T) and B3LYP levels of theory. Later, the accurate three-dimensional, near-equilibrium potential energy and dipole moment functions for both the ground and the first electronic excited states of HS2 were constructed by Peterson et al.[17] by using highly correlated coupled cluster methods with explicit basis set extrapolation. Song et al.[18] reported an accurate double many-body expansion (DMBE) potential energy surface (PES) for ground-state HS2 based on ab initio energies extrapolated to the complete basis set limit. Quite recently, Qin et al.[7] calculated the equilibrium geometry of the first excited electronic state of HS2 by time-dependent density functional theory using the hybrid functional B3LYP with the aug-cc-pVQZ (AVQZ) basis set.
The vibrational frequencies of the ground state (X2A″) and the excited state (A2A′) of HS2 were investigated by Holstein et al.[19] By studying the microwave spectroscopy of the ground electronic state of HS2, Yamanoto and Saito[20] reported the lowest energy structure. Ashworth et al.[21] first observed the far infrared laser magnetic resonance (LMR) spectrum of the ground state of HS2. Subsequently, Isoniemi et al.[22] investigated more refined structural information of HS2 radical in Ar matrix following the 266 nm photolysis of H2S2. The S–H stretching and the S–S–H bending modes were observed at 2463 cm−1 and 903 cm−1, respectively. Ashworth and Fink[23] analyzed the X–A band system through high-resolution Fourier-transform measurements of the chemiluminescence spectra of both HS2 and DS2. Entfellner and Boesl[24] presented the photodetachment-photoelectron spectra of the ground and the first excited states of HS2 and DS2 with a conventional time of flight photoelectron spectrometry.
Indeed, as far as we are aware, there is no work toward obtaining a global PES for the first electronic excited state of HS2(A2A′) reported, which would be of interest to the investigations of the H(2S) + S2(a1Δg)→ SH(X2Π) + S(3P) reaction. Thus, the major goal of the present work is to obtain a high quality global PES of HS2(A2A′) based on the DMBE[25–28] methodology.
This paper is organized as follows. Section 2 describes the ab initio calculations employed in the present work. Section 3 presents a brief description of the analytical DMBE formalism. Then the major topographical features of the HS2(A2A′) PES are discussed in Section 4. The concluding remarks are summarized in Section 5.
The ab initio calculations have been carried out at the multi-reference configuration interaction level[29,30] of theory, including the Davidson correction [MRCI(Q)],[31] using the full valence complete-active-space self-consistent field (CASSCF)[32] wave function as the reference. The AVQZ basis set of Dunning[33,34] is employed, and the ab initio calculations are performed using the Molpro 2012 package.[35] A grid of 2767 ab initio points is chosen to map the PES over the H–S2 region defined by 2.5 ≤ RS2/a0 ≤ 5.5, 0.2 ≤ rH−S2/a0 ≤ 15, 0° ≤ γ/≤ 90°, and for the S–SH interactions, 2.0 ≤ RSH/a0 ≤ 4.0, 0.6 ≤ rS−SH/a0 ≤ 15, 0° ≤ γ/≤ 180°, where R, r, and γ are the atom–diatom Jacobi coordinates. The Cs point group symmetry is employed in the ab initio calculations, which holds two irreducible representations A′ and A″. For HS2(A2A′), seven A′ and two A″ symmetry molecular orbitals (MOs) are determined as the active space, amounting to 214 (136A′ + 78A″) configuration state functions. The MOs which correlate with the 3s and 3p atomic orbitals of each S and the 1s orbital of the H atom are utilized to determine the valence space.
The ab initio energies calculated in this way have been subsequently corrected using the double many-body expansion-scaled external correlation method (DMBE-SEC)[36] to account for the excitations beyond singles and doubles, most importantly, for incompleteness of the basis set. According to the DMBE-SEC scheme, the total DMBE-SEC interaction energy is written as
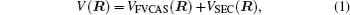
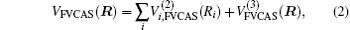
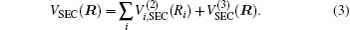
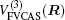
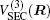
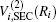
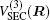
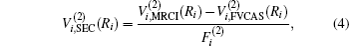
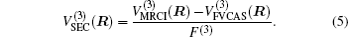
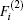
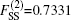
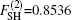
In the framework of DMBE methodology, the single-sheeted PES can be written as
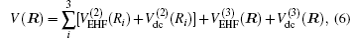
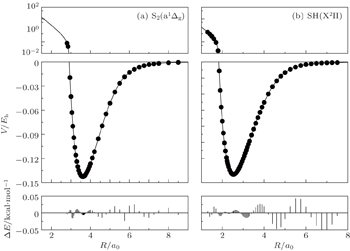
The diatomic potential energy curves (PECs) of S2(a 1Δg) and SH(X2Π), which show the correct behaviors at the asymptotic limits R→ 0 and R → ∞, have been modeled using the EHF approximate correlation energy method.[40,41]
The EHF-type energy term is written as

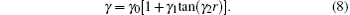
In turn, the dc energy term is written as[42]
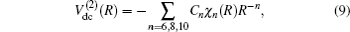
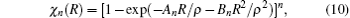
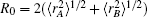
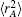
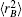
As usual,[44,45] the three-body dc energy term is written in the following functional form:
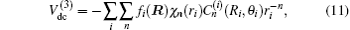
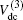
For a given triatomic geometry, by removing the sum of the two-body energy terms from the corresponding interaction energies, one obtains the total three-body energy. Then by subtracting the three-body dc contribution from the total three-body energy, the three-body EHF energy contribution is obtained. It can be modeled in the three-body distributed-polynomial[47] form
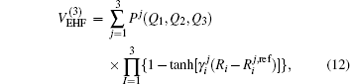
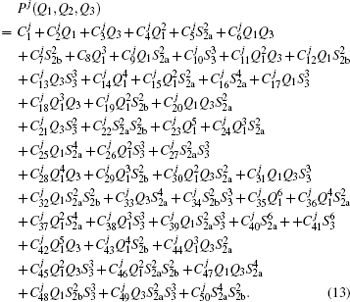
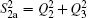
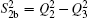
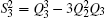
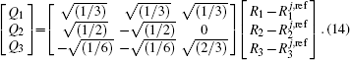
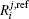
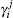
| Table 1. Root-mean-square deviations (in kcal·mol−1) of the PES. . |
The fitted potential energy curves of S2(a 1Δg) and SH(X2Π) and the deviations from the MRCI(Q)/AVQZ energies are displayed in Fig.
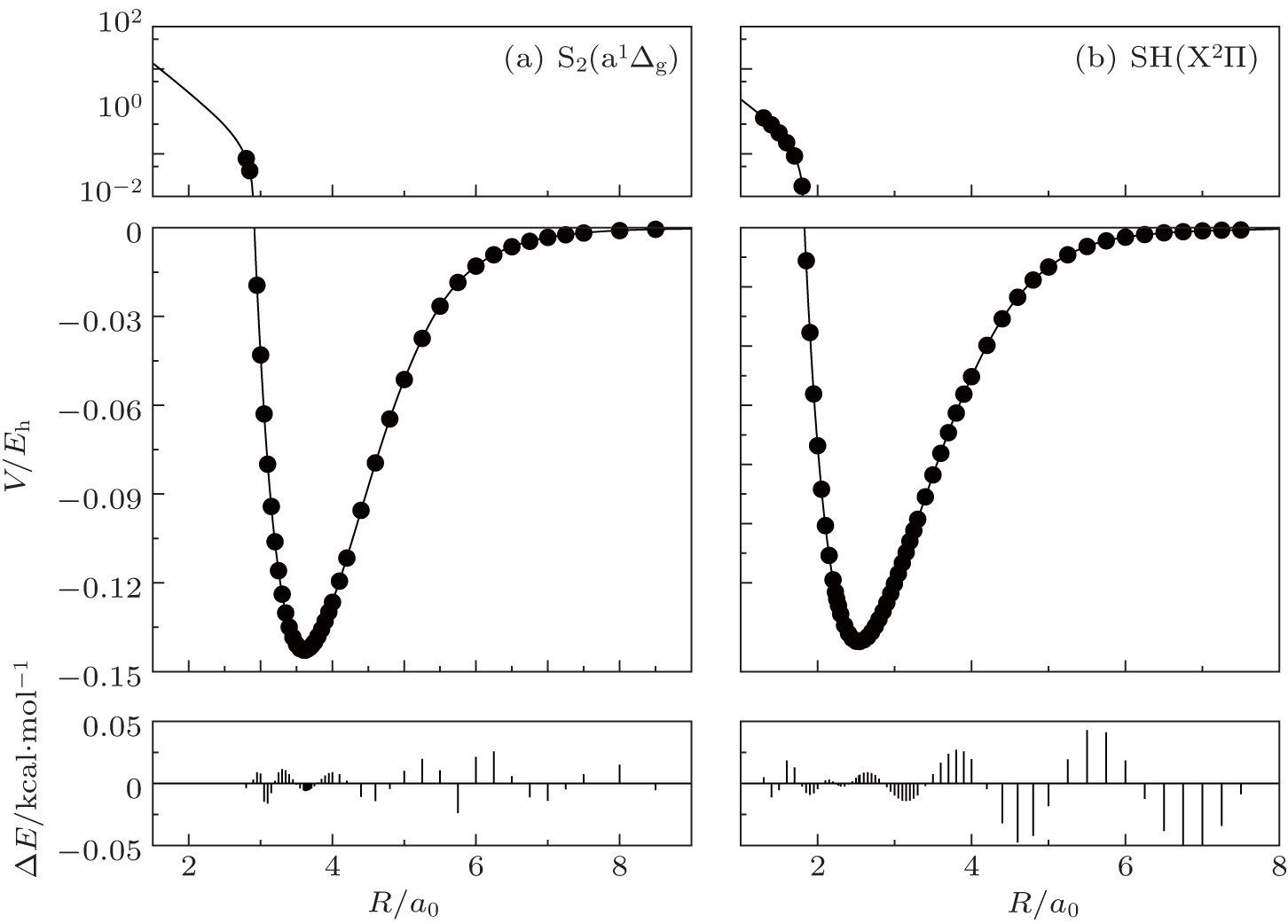 | Fig. 1. Potential energy curves of (a) S2(a 1Δg) and (b) SH(X2Π), and the differences between the fitted PECs and the ab initio points. |
| Table 2. Spectroscopic constants of SH and S2 diatoms, Re is in the units of a.u., De is in the units of kcal · mol−1, and ωe, ωexe, αe, and βe are in the units of cm−1. . |
Figures
 | Fig. 2. Contour plots of HS2 (A2A′) potential energies for the four H–S–S angles (a) 0°, (b) 45°, (c) 90°, and (d) 135°. Contours are equally spaced by 0.006Eh, starting from −0.1668Eh in panel (a), −0.1977Eh inpanel (b), −0.2294Eh in panel (c), and −0.2006Eh in panel (d). |
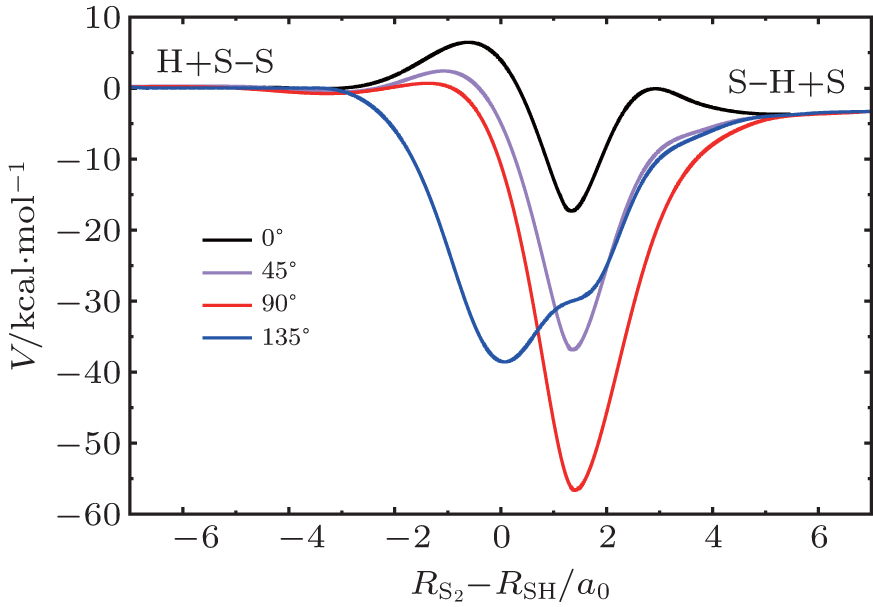 | Fig. 3. Minimum energy paths for HS2(A2A′) PES at four H–S–S angles. |
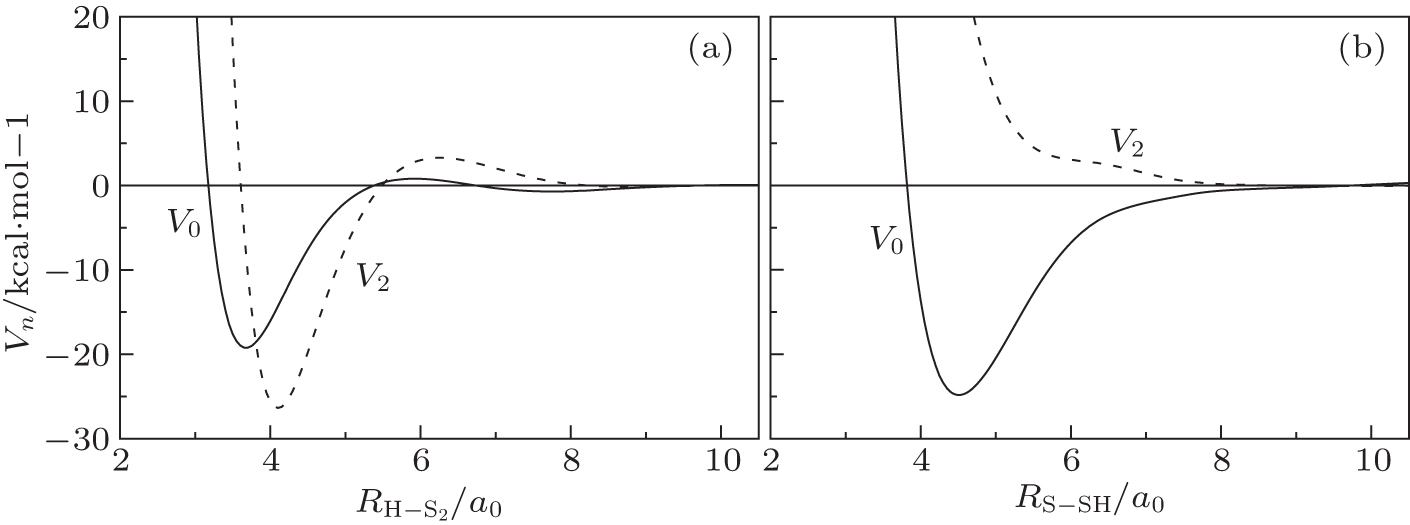 | Fig. 4. The spherically averaged isotropic (V0) and leading anisotropic potentials (V2): (a) for H + S2 interaction, with RSS = 3.6079a0; (b) for S + SH interaction, with RSH = 2.5337a0. |
| Table 3. Properties of stationary points on the fitted HS2(A2A′) PES (harmonic frequencies in cm−1). . |
The contour maps for the H(2S) + S2(a1Δg) → SH(X 2Π) + S(3P) reaction in internal coordinates at four H–S–S angles (∠HSS = 0°, 45°, 90°, and 135°) are shown in Fig.
Figure

A global accurate PES is reported for HS2(A2A′) by fitting the MRCI(Q)/AVQZ energies which are scaled by the DMBE-SEC method. The resulting PES is realistic over the entire configuration space. The various topographical features of the current DMBE/SEC PES have been examined in detail and compared with the previous theoretical and experimental results. The properties, including geometries, energies, and vibrational frequencies, have been characterized on the current HS2(A2A′) DMEB-SEC PES. The results show good agreement with the previous high quality theoretical studies and the available experiment data. Thus, the PES constructed here can be used for dynamic studies of reactions and also as a building block for the PESs of larger molecular systems containing the HS2 fragment.
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 | |
| 22 | |
| 23 | |
| 24 | |
| 25 | |
| 26 | |
| 27 | |
| 28 | |
| 29 | |
| 30 | |
| 31 | |
| 32 | |
| 33 | |
| 34 | |
| 35 | |
| 36 | |
| 37 | |
| 38 | |
| 39 | |
| 40 | |
| 41 | |
| 42 | |
| 43 | |
| 44 | |
| 45 | |
| 46 | |
| 47 | |
| 48 | |
| 49 | |
| 50 | |
| 51 | |
| 52 | |
| 53 | |
| 54 | |
| 55 | |
| 56 | |
| 57 | |
| 58 | |
| 59 | |
| 60 |