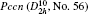
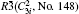
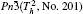
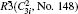
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Electronic structures of efficient MBiO3 (M = Li, Na, K, Ag) photocatalyst
[Zhou Wen-Liu, Zhao Zong-Yan†,  ]
]
]
|
|


† Corresponding author. E-mail:
Project supported by the National Natural Science Foundation of China (Grant No. 21473082).
In order to deepen the understanding of the relationship between fundamental properties (including: microstructure and composition) and photocatalytic performance, four bismuthate compounds, including: LiBiO3, NaBiO3, KBiO3, and AgBiO3, are regarded as research examples in the present work, because they have particular crystal structures and similar compositions. Using density functional theory calculations, their structural, electronic, and optical properties are investigated and compared systematically. First of all, the calculated results of crystal structures and optical properties are in agreement with available published experimental data. Based on the calculated results, it is found that the tunneled or layered micro-structural properties lead to the stronger interaction between bismuth and oxygen, and the weaker interaction between alkaline-earth metal and [BiO6] octahedron, resulting in the feature of multi-band gaps in the cases of LiBiO3, NaBiO3, and KBiO3. This conclusion is supported by the case of AgBiO3, in which the feature of multi-band gaps disappears, due to the stronger interaction between the noble metal and [BiO6] octahedron. These properties have significant advantages in the photocatalytic performance: absorbing low energy photons, rapidly transferring energy carriers. Furthermore, the features of electronic structures of bismuthate compounds are well reflected by the absorption spectra, which could be confirmed by experimental measurements in practice. Combined with the calculated results, it could be considered that the crystal structures and compositions of the photocatalyst determine the electronic structures and optical properties, and subsequently determine the corresponding photocatalytic performance. Thus, a novel Bi-based photocatalyst driven by visible-light could be designed by utilizing specific compositions to form favorable electronic structures or specific micro-structures to form a beneficial channel for energy carriers.
With rapid economy development, people are facing more and more severe challenges of energy shortage and environmental deterioration, which are becoming increasingly prominent and attract more and more attention. Photocatalysis technology can provide effective solutions to address these challenges.[1,2] At present, most research on photocatalysis focuses on the modifications for TiO2,[3] because it is the prototype for the development of a novel photocatalyst. In the 1970s, Fujishima and Honda found electrochemical photolysis of water at the TiO2 electrode under UV-light irradiation,[4] which opened a new era of heterogeneous photocatalysis. However, as a wide band gap (3.0–3.2 eV) semiconductor, TiO2 only responds to UV-light, and thus its solar energy utilization efficiency is very low. At the same time, the photo-generated electron–hole pairs can recombine very easily, resulting in its low quantum conversion efficiency. Although researchers attempt to improve its photocatalytic performance by using modification methods (such as, impurity doping, noble metal loading, hetero-structure constructing, and dye sanitizing, etc.),[5,6] it is difficult for its performance to achieve the requirements of industrialized application in the short term. Therefore, to develop a novel efficient photocatalyst driven by visible-light is a major trend in the recent research of photocatalysis.
In recent years, the Bi-based photocatalyst has attracted wide attention because of the interesting photocatalytic performance under visible-light irradiation, and the diversity and flexibility of its structures and properties. It has been found that many Bi-based compound semiconductors have narrow band gaps and present efficient photocatalytic activity, resulting from the hybridized valence band between O-2p states and Bi-6s states. These results indicated that Bi-based compounds and composites have become an important family of photocatalysts driven by visible-light.[7–9] In particular, bismuthates (MBiO3) contain Bi5+ with 6s empty orbital, which contributes to both the top of the valence band and the bottom of the conduction band. This feature can vary the band edge positions and narrow the band gap, leading to the relatively high photocatalytic activity under visible-light irradiation.[10] For example, Ramachandran et al. evaluated the photocatalytic activity of KBiO3 and LiBiO3 for the degradation of anionic and cationic dyes under UV and solar radiations, and observed that the dye structure determines the effectiveness of photocatalytic reactions.[11] NaBiO3 also exhibits much higher activity compared with BiVO4 and N-doped TiO2 for the decomposition of methylene blue under visible light. Kako et al. considered that the attractive performance of NaBiO3 is ascribed to its band structure where the CB consists of hybridized Na-3s and O-2p orbitals that exhibit large dispersion with high mobility for photo-excited electrons.[12]
Although bismuthates exhibit high photocatalytic activity, their fundamental photophysical properties have not been systematically studied and compared. In order to deepen the understanding of the relationship between fundamental properties (including: microstructure and composition) and photocatalytic performance, four bismuthate compounds MBiO3 (M = Li, Na, K, Ag) are regarded as research examples in the present work, because they have particular crystal structures and similar compositions. (i) LiBiO3 and KBiO3 have tunneled structures, while NaBiO3 and AgBiO3 have layered structures. (ii) Li, Na, and K belong to the elements of alkaline-earth metals that have similar features. (iii) Although Na and Ag belong to different periodic table groups, NaBiO3 and AgBiO3 have the exact same crystal structure. In short, the similarities and differences between these four compounds have provided an opportunity to thoroughly investigate the relationship between the fundamental properties and the photocatalytic performance of bismuthates. The previous experimental observations about these MBiO3 photocatalysts were independently reported by different research groups. Since their synthesis processing, experimental condition, and evaluation criteria are not completely consistent, so their experimental results cannot directly be compared to achieve this purpose. Different from the experimental study, the atomic-scale theoretical calculation can obtain comparable results for different materials based on the unified computational method and platform.[13–15] Hence, in this paper, we used density functional theory (DFT) to calculate the crystal structures, electronic structures, and optical properties of MBiO3 (M = Li, Na, K, Ag). Based on the calculated results, the underlying photocatalytic mechanism of MBiO3 and the corresponding photocatalytic performance are analyzed. Our calculated results would be used as a reference for the development of efficient Bi-based photocatalyst driven by visible-light using specific compositions to form favorable electronic structures or specific micro-structures to form a beneficial channel for energy carriers.
In the present work, the original crystal models are taken from the Inorganic Crystal Structure Database (ICSD). All of the DFT calculations were carried out by using Cambridge Serial Total Energy Package (CASTEP) codes that is interpolated in the package of Materials Studio 5.5,[16] which is a state-of-the-art quantum mechanics-based program designed specifically for solid-state materials science. According to the Born–Oppenheimer approximation and Hartree–Fock approximation, the core electrons (Bi: [Xe], O: [He], Li: [He], Na: [Ne], K: [Ar], Ag: [Kr]) were treated with the ultra soft pseudo-potential, while the exchange–correlation effects of valence electrons (Bi: 6s26p3, O: 2s22p4, Li: 1s22s1, Na: 2s22p63s1, K: 3s23p64s1, Ag: 4d105s1) were described by the revised Perdew–Burke–Ernzerh for solid (PBEsol) of generalized gradient approximation (GGA).[17] It is well known that standard GGA method underestimates the band gap of semiconductor by ∼ 50%,[18] consequently the calculated results could not be compared with and confirmed by the experimental measurements directly. In order to overcome the shortcoming of GGA and obtain accurate electronic structures, the GGA+U method was adopted in the present work, and the values of Ueff were set as follows: 2 eV for the p states of Bi, Na, K, and O; 3 eV for Ag-d states. The values of Ueff are determined by the comparison between calculated band gaps and the corresponding experimental results, according to the repeated test. In the present work, we set the same values of Ueff for Bi and O in all compounds.
An energy cutoff of 380–410 eV was used to expand the Kohn–Sham wave functions. The Monkhorst–Pack scheme k-points grid sampling was set as 2×3×1–3×3×3 for the irreducible Brillouin zone. A 40×40×40–60×60×60 mesh was used for fast Fourier transformation. We use Broyden–Fletcher–Goldfarb–Shanno (BFGS) scheme[19] as the minimization algorithm, in which all atomic positions and lattice parameters are optimized. Its convergence criteria were set as follows: the force on the atoms was less than 0.01 eV/Å, the stress on the atoms was less than 0.02 GPa, the atomic displacement was less than 5×10−4 Å, and the energy change per atom was less than 5×10−6 eV. Based on the optimized crystal structures, the electronic structures and optical properties were then calculated.
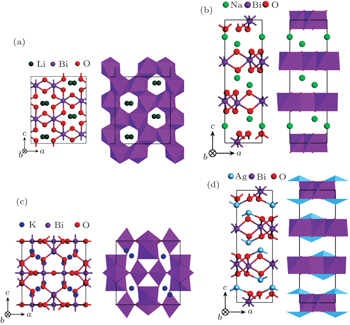
Four crystal structures of bismuthates are illustrated in Fig. 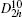
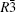
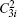
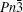
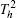
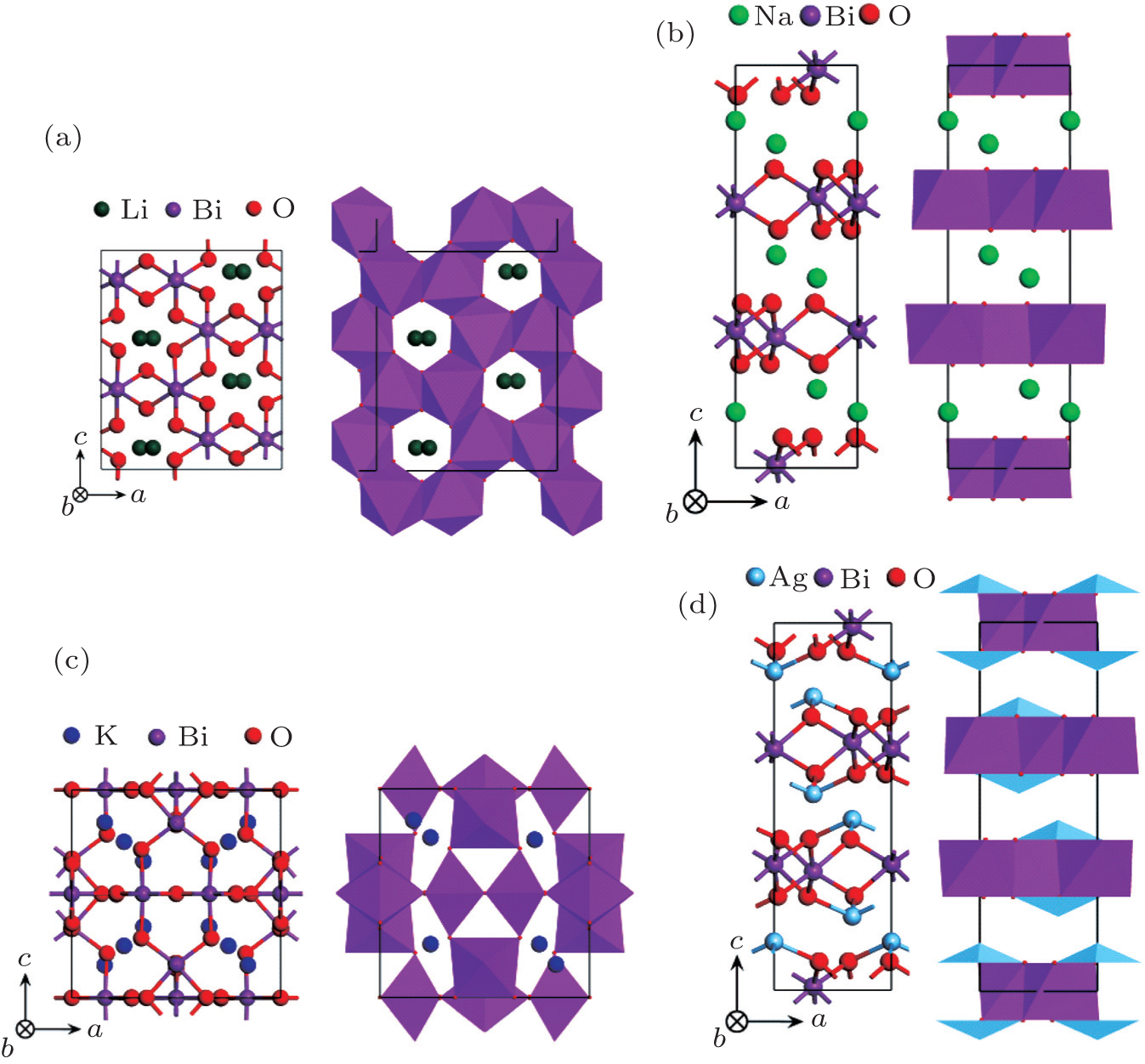 | Fig. 1. The crystal structure models of four bismuthate compounds: (a) LiBiO3, (b) NaBiO3, (c) KBiO3, and (d) AgBiO3. |
All the crystal lattice parameters for these four bismuthates are tabulated in Table 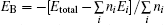
| Table 1. The lattice constants and parameters, and the atomic average net charge from Mulliken population analysis of bismuthate compounds. . |
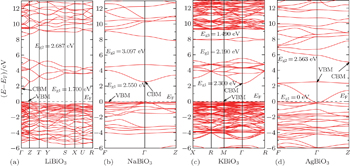
The band structures of these four bismuthates are illustrated in Fig.
In the case of AgBiO3, the highest VB and the lowest CB contact together, indicating that the minimum value of band gap is 0 eV. Above the lowest CB, one could see another forbidden band with an indirect pseudo-band gap of 2.563 eV, in which the pseudo-VBM/CBM is located at Γ/Z k-point. The band-gap value of AgBiO3 is a very controversial issue. The sample color of AgBiO3 is black,[25,26] implying its band gap is very small. Based on the UV–vis spectra, Takei et al. estimated its band gap is 0.87 eV,[26] while Yu et al. estimated its band gap is 2.5 eV.[27] On the contrary, using different calculation methods (linear muffin-tin orbital method and discrete-variational X α molecular orbital method), Mizoguchi et al.[25] and Takei et al.[26] independently obtained the band gap of AgBiO3 is 0 eV. Carefully comparing the experimental measurements with theoretical calculations, we considered that the band gap of AgBiO3 should be determined as 0 eV, and the experimental measurements might come from the electron transition deviated from Γ k-point or the electron transition between pseudo-VBM and pseudo-CBM in the second forbidden band.
The parameters of band structures of these four bismuthate compounds are listed in Table
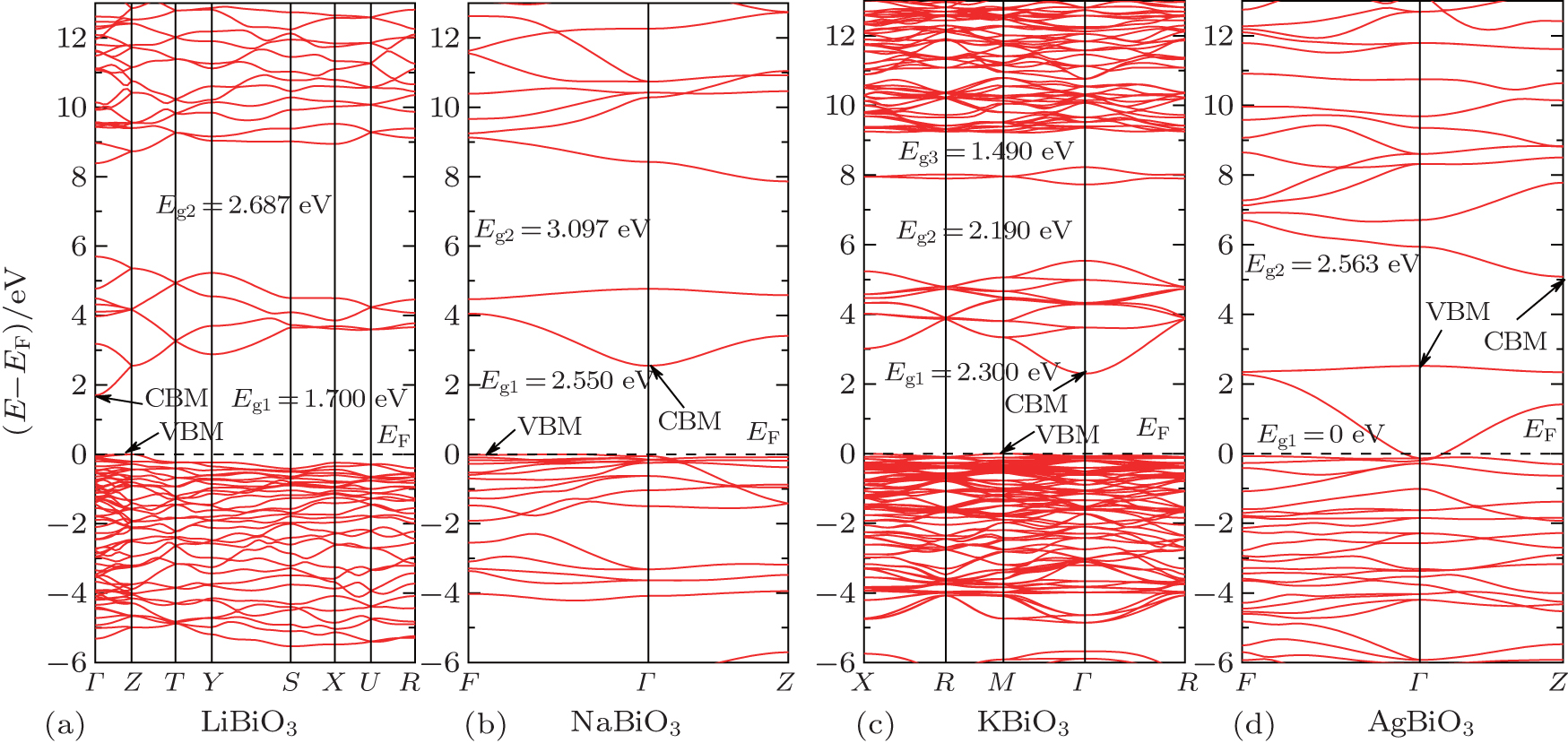 | Fig. 2. The calculated band structures of four bismuthate compounds. (a) LiBiO3, (b) NaBiO3, (c) KBiO3, and (d) AgBiO3. |
| Table 2. The parameters of band structures of bismuthate compounds, the energy unit is eV. . |
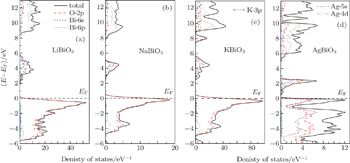
To analyze the chemical bonding information, the total and partial density of states (DOS) of these four bismuthate compounds are plotted and compared in Fig.
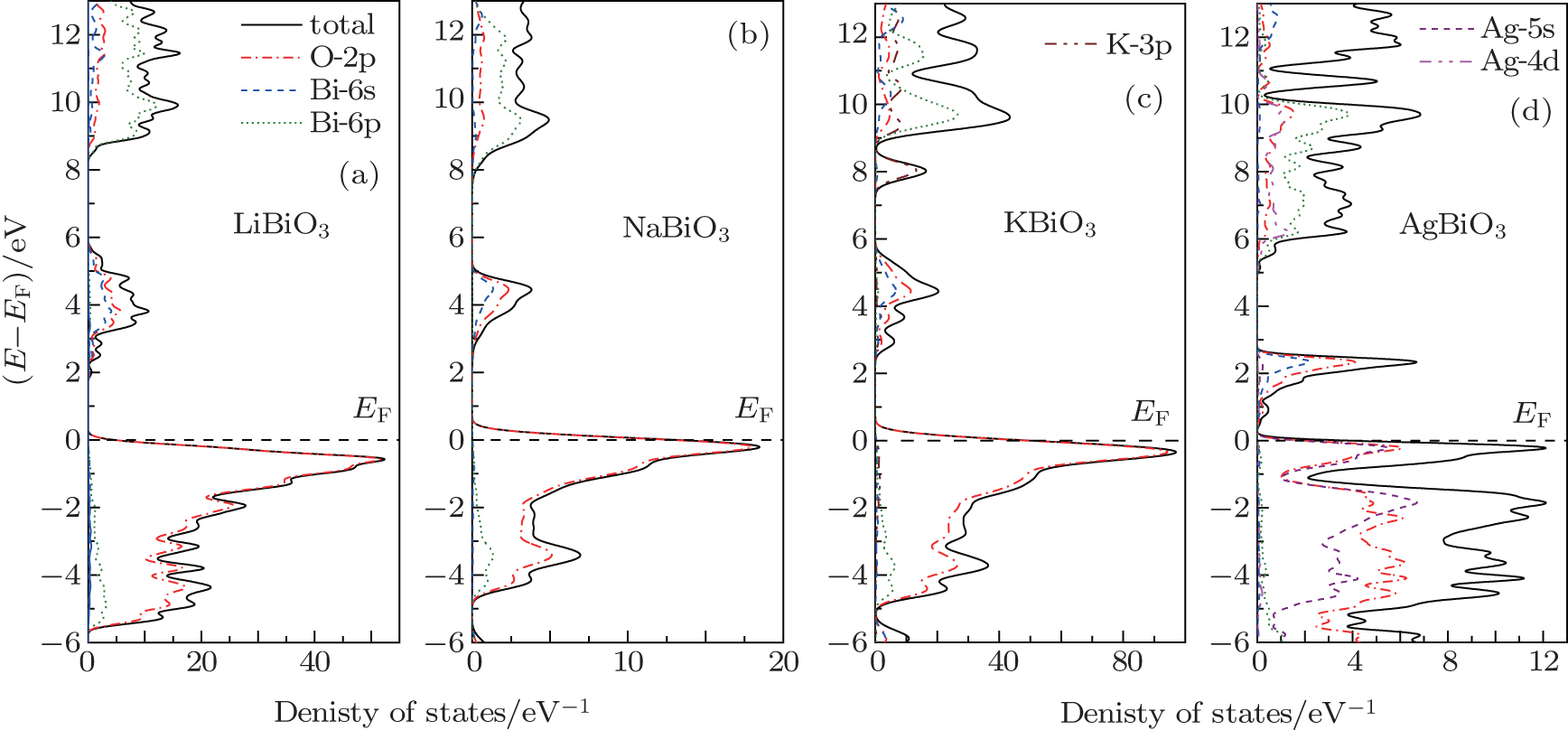 | Fig. 3. The calculated density of states of four bismuthate compounds. (a) LiBiO3, (b) NaBiO3, (c) KBiO3, and (d) AgBiO3. |
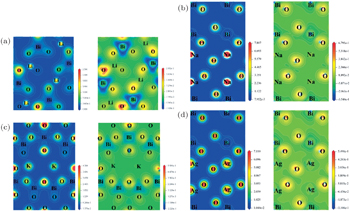
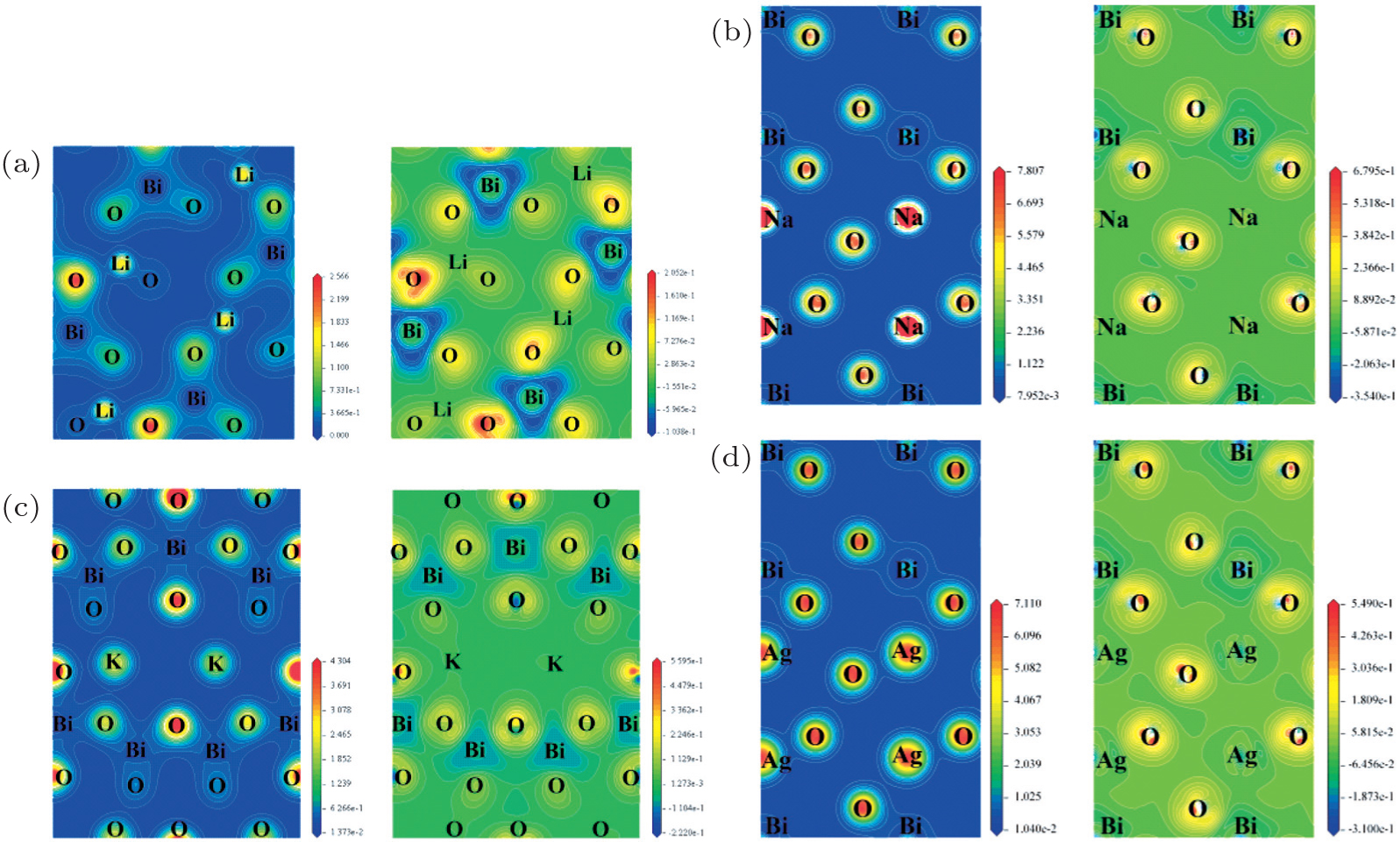 | Fig. 4. The calculated contour maps of electron densities (right) and electron difference densities (left) of four bismuthate compounds. (a) LiBiO3, (b) NaBiO3, (c) KBiO3, and (d) AgBiO3. |
The contour maps of electron densities and electron difference densities of these four bismuthate compounds are plotted and compared in Fig.
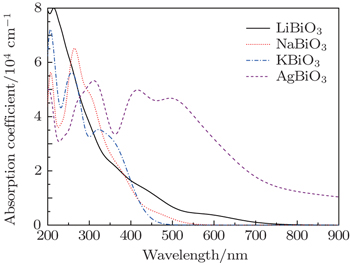
Figure
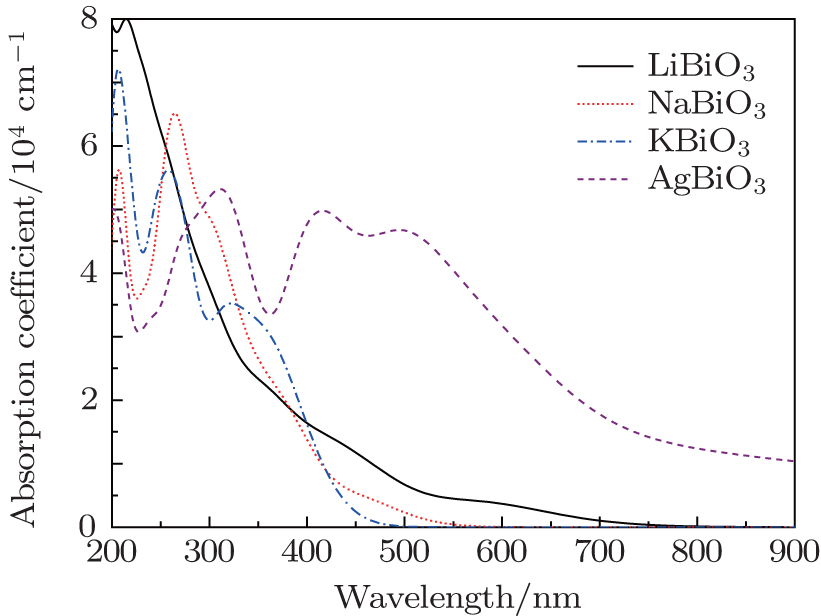 | Fig. 5. The calculated absorption spectra of four bismuthate compounds by polycrystalline models. |
However the absorption spectra calculated by DFT calculations are different from the experimental results, for example in the case of AgBiO3. The differences may be because only a straightforward transition from occupied energy bands (VB) to unoccupied energy bands (CBL and CBU) is considered in DFT calculations, while in practice, owing to the semi-metallic features, the electron transition from CBL to CBU could occur. However, their underlying mechanisms are identical to that determined by the electron transfer from the occupied energy bands to the unoccupied energy bands by photon excitation. Taking the experimental literature Ref. [27] as an example, the authors determined the optical band gap, based on the absorption edge in the range of 400–600 nm, which is corresponding to the electron transition from VB → CBU in Fig.
The crystal structures, electronic structures, and optical properties of four bismuthate compounds (including: LiBiO3, NaBiO3, KBiO3, and AgBiO3) have been calculated by density functional theory. LiBiO3 and KBiO3 have tunneled structures, and NaBiO3 has a layered structure. Thus, Li+, Na+, or K+ ions may easily transfer in the tunnel or the layer space to carry the solar energy, owing to the small ionic radii and the weak interaction with [BiO6] octahedrons. Although AgBiO3 has an identical layered structure to NaBiO3, the strong interaction between Ag atoms and O atoms, and the large radius of Ag+, result in the Ag+ ions being unable to transfer freely and form [AgO3] tetrahedron. The tunneled and layered micro-structures make these four bismuthate compounds have multi-band gaps, namely their bands near the fundamental band gap are located and separated. Also, these features of electronic structure are reflected by the absorption spectra, which could be confirmed by the experimental measurements. Based on the calculated results, it could be considered that the crystal structures and compositions of photocatalyst determine the electronic structures and optical properties, and subsequently determine the corresponding photocatalytic performance. Furthermore, the present work also presents some open issues that are worthy of in-depth investigation, such as: the transfer behavior of ions with small radii in the tunneled or layered photocatalyst, and the effects of the semi-metallic feature on photocatalytic performance.
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 | |
| 22 | |
| 23 | |
| 24 | |
| 25 | |
| 26 | |
| 27 |